User login
Vaginal ring lowers HIV transmissions in two trials
BOSTON – A vaginal ring infused with the antiretroviral drug dapivirine is effective and safe for reducing HIV-1 infections in high-risk women; at least in women older than 21 who use it as intended, results of two large randomized trials suggest.
In the ASPIRE Trial, conducted from August 2012 through June 2015 in Malawi, South Africa, Uganda, and Zimbabwe, there were 71 HIV infections among 1,313 women assigned to use the dapivirine ring, compared with 97 among 1,316 women assigned to a placebo, a difference that translated into a 27% reduction in infections.
In the Ring Study, conducted in South Africa and Uganda, the drug-infused ring reduced infections in the overall population by 31%, compared with a placebo ring, reported Dr. Annalene Nel from the International Partnership for Microbicides in Paarl, South Africa.

“This is the first demonstration of a sustained-release approach for antiretroviral prevention, and HIV protection in this study, not surprisingly, was greater with greater adherence,” said ASPIRE lead investigator Dr. Jared M. Baeten of the University of Washington, Seattle, at a media briefing at the Conference on Retroviruses and Opportunistic Infections.
“The data from both studies show that the ring must be used consistently to achieve protection, and that protection can be achieved with consistent use,” Dr. Nel said at the briefing.
The ASPIRE results are also published online in the New England Journal of Medicine (2016 Feb 22. doi: 10.1056/NEJMoa1506110).
In both trials, the ring offered little or no significant protection for women aged 18-21. In ASPIRE, there was no benefit for the dapivirine ring in this age group, and in Ring, infections were 15% lower among younger women who received the active ring, compared with placebo.

The ASPIRE investigators noted that younger women tended to have lower objective markers of adherence, such as dapivirine levels in plasma and in used rings, suggesting that poor adherence could partly explain the lack of benefit in this age group. They also suggest that “the genital tract of women in this age group may be more susceptible to HIV-1 infection and potentially also more difficult to protect with antiretroviral prophylaxis strategies.”
Trial design
In each trial, sexually active, HIV-negative women aged 18-45 were enrolled and randomly assigned to receive a vaginal ring containing 25 mg of dapivirine, a non-nucleoside HIV-1 reverse transcriptase inhibitor, or a placebo ring. The rings were replaced every 4 weeks. In ASPIRE, the randomization was on a 1:1 basis; in the Ring study, the randomization was 2:1, with twice as many women receiving the active ring.
Participants had monthly follow-up visits that included serologic testing, safety monitoring, and adherence counseling. Median follow-up in ASPIRE was 1.6 years. The Ring study was halted early at the recommendation of the data safety monitoring board because of a higher-than-expected incidence of HIV infections.
ASPIRE study
In addition to the overall 27% reduction in infections seen in ASPIRE, the investigators found that when they excluded data from two sites with lower rates of retention and adherence, the ring reduced infections by 37%.
A post hoc analysis showed significantly better protection for women over age 21 (P less than .001), but not for younger women (P = .45), a difference that correlated with reduced adherence.
The researchers noted that among women who acquired HIV, the rates of adverse medical events and resistance to antiretroviral agents were similar between the active ring and placebo groups.
Ring study
As of Oct. 16, 2015 – the data cutoff point for RING – there were 2,805 person-years of follow-up, and 761 women had completed 2 years of follow-up. There were a total of 133 post-randomization HIV-1 infections, 77 of which occurred among women assigned to the dapivirine ring, translating into an incidence of 4.08 per 100 person-years, and 56 of which occurred among women assigned to placebo, for an incidence of 6.10 per 100 person-years.
The overall reduction in infections in the active ring group in the Ring study relative to placebo was 31% (P = .0401). In a subgroup analysis of women over age 21, the dapivirine ring reduced infections by 37%.
The most common ring-related adverse events in this trial were abnormal uterine bleeding, pelvic discomfort and/or pain, suprapubic pain, and application site pain. Adverse-event rates, including product-related events, urogenital events, serious adverse events and death, were similar between the treatment arms.
Based on the results of the two trials, the International Partnership for Microbicides is planning an open-label extension study for the Ring cohort and submission of data for regulatory approval of the device.
The ASPIRE study was funded by the National Institutes of Health. The Ring Study was funded by the Bill and Melinda Gates Foundation, the U.S. President’s Emergency Plan for AIDS Relief, the U.S. Agency for International Development, and several European governments and organizations, and was conducted by the International Partnership for Microbicides.
Dr. Baeten and Dr. Nel reported having no financial disclosures.
BOSTON – A vaginal ring infused with the antiretroviral drug dapivirine is effective and safe for reducing HIV-1 infections in high-risk women; at least in women older than 21 who use it as intended, results of two large randomized trials suggest.
In the ASPIRE Trial, conducted from August 2012 through June 2015 in Malawi, South Africa, Uganda, and Zimbabwe, there were 71 HIV infections among 1,313 women assigned to use the dapivirine ring, compared with 97 among 1,316 women assigned to a placebo, a difference that translated into a 27% reduction in infections.
In the Ring Study, conducted in South Africa and Uganda, the drug-infused ring reduced infections in the overall population by 31%, compared with a placebo ring, reported Dr. Annalene Nel from the International Partnership for Microbicides in Paarl, South Africa.
“This is the first demonstration of a sustained-release approach for antiretroviral prevention, and HIV protection in this study, not surprisingly, was greater with greater adherence,” said ASPIRE lead investigator Dr. Jared M. Baeten of the University of Washington, Seattle, at a media briefing at the Conference on Retroviruses and Opportunistic Infections.
“The data from both studies show that the ring must be used consistently to achieve protection, and that protection can be achieved with consistent use,” Dr. Nel said at the briefing.
The ASPIRE results are also published online in the New England Journal of Medicine (2016 Feb 22. doi: 10.1056/NEJMoa1506110).
In both trials, the ring offered little or no significant protection for women aged 18-21. In ASPIRE, there was no benefit for the dapivirine ring in this age group, and in Ring, infections were 15% lower among younger women who received the active ring, compared with placebo.
The ASPIRE investigators noted that younger women tended to have lower objective markers of adherence, such as dapivirine levels in plasma and in used rings, suggesting that poor adherence could partly explain the lack of benefit in this age group. They also suggest that “the genital tract of women in this age group may be more susceptible to HIV-1 infection and potentially also more difficult to protect with antiretroviral prophylaxis strategies.”
Trial design
In each trial, sexually active, HIV-negative women aged 18-45 were enrolled and randomly assigned to receive a vaginal ring containing 25 mg of dapivirine, a non-nucleoside HIV-1 reverse transcriptase inhibitor, or a placebo ring. The rings were replaced every 4 weeks. In ASPIRE, the randomization was on a 1:1 basis; in the Ring study, the randomization was 2:1, with twice as many women receiving the active ring.
Participants had monthly follow-up visits that included serologic testing, safety monitoring, and adherence counseling. Median follow-up in ASPIRE was 1.6 years. The Ring study was halted early at the recommendation of the data safety monitoring board because of a higher-than-expected incidence of HIV infections.
ASPIRE study
In addition to the overall 27% reduction in infections seen in ASPIRE, the investigators found that when they excluded data from two sites with lower rates of retention and adherence, the ring reduced infections by 37%.
A post hoc analysis showed significantly better protection for women over age 21 (P less than .001), but not for younger women (P = .45), a difference that correlated with reduced adherence.
The researchers noted that among women who acquired HIV, the rates of adverse medical events and resistance to antiretroviral agents were similar between the active ring and placebo groups.
Ring study
As of Oct. 16, 2015 – the data cutoff point for RING – there were 2,805 person-years of follow-up, and 761 women had completed 2 years of follow-up. There were a total of 133 post-randomization HIV-1 infections, 77 of which occurred among women assigned to the dapivirine ring, translating into an incidence of 4.08 per 100 person-years, and 56 of which occurred among women assigned to placebo, for an incidence of 6.10 per 100 person-years.
The overall reduction in infections in the active ring group in the Ring study relative to placebo was 31% (P = .0401). In a subgroup analysis of women over age 21, the dapivirine ring reduced infections by 37%.
The most common ring-related adverse events in this trial were abnormal uterine bleeding, pelvic discomfort and/or pain, suprapubic pain, and application site pain. Adverse-event rates, including product-related events, urogenital events, serious adverse events and death, were similar between the treatment arms.
Based on the results of the two trials, the International Partnership for Microbicides is planning an open-label extension study for the Ring cohort and submission of data for regulatory approval of the device.
The ASPIRE study was funded by the National Institutes of Health. The Ring Study was funded by the Bill and Melinda Gates Foundation, the U.S. President’s Emergency Plan for AIDS Relief, the U.S. Agency for International Development, and several European governments and organizations, and was conducted by the International Partnership for Microbicides.
Dr. Baeten and Dr. Nel reported having no financial disclosures.
BOSTON – A vaginal ring infused with the antiretroviral drug dapivirine is effective and safe for reducing HIV-1 infections in high-risk women; at least in women older than 21 who use it as intended, results of two large randomized trials suggest.
In the ASPIRE Trial, conducted from August 2012 through June 2015 in Malawi, South Africa, Uganda, and Zimbabwe, there were 71 HIV infections among 1,313 women assigned to use the dapivirine ring, compared with 97 among 1,316 women assigned to a placebo, a difference that translated into a 27% reduction in infections.
In the Ring Study, conducted in South Africa and Uganda, the drug-infused ring reduced infections in the overall population by 31%, compared with a placebo ring, reported Dr. Annalene Nel from the International Partnership for Microbicides in Paarl, South Africa.
“This is the first demonstration of a sustained-release approach for antiretroviral prevention, and HIV protection in this study, not surprisingly, was greater with greater adherence,” said ASPIRE lead investigator Dr. Jared M. Baeten of the University of Washington, Seattle, at a media briefing at the Conference on Retroviruses and Opportunistic Infections.
“The data from both studies show that the ring must be used consistently to achieve protection, and that protection can be achieved with consistent use,” Dr. Nel said at the briefing.
The ASPIRE results are also published online in the New England Journal of Medicine (2016 Feb 22. doi: 10.1056/NEJMoa1506110).
In both trials, the ring offered little or no significant protection for women aged 18-21. In ASPIRE, there was no benefit for the dapivirine ring in this age group, and in Ring, infections were 15% lower among younger women who received the active ring, compared with placebo.
The ASPIRE investigators noted that younger women tended to have lower objective markers of adherence, such as dapivirine levels in plasma and in used rings, suggesting that poor adherence could partly explain the lack of benefit in this age group. They also suggest that “the genital tract of women in this age group may be more susceptible to HIV-1 infection and potentially also more difficult to protect with antiretroviral prophylaxis strategies.”
Trial design
In each trial, sexually active, HIV-negative women aged 18-45 were enrolled and randomly assigned to receive a vaginal ring containing 25 mg of dapivirine, a non-nucleoside HIV-1 reverse transcriptase inhibitor, or a placebo ring. The rings were replaced every 4 weeks. In ASPIRE, the randomization was on a 1:1 basis; in the Ring study, the randomization was 2:1, with twice as many women receiving the active ring.
Participants had monthly follow-up visits that included serologic testing, safety monitoring, and adherence counseling. Median follow-up in ASPIRE was 1.6 years. The Ring study was halted early at the recommendation of the data safety monitoring board because of a higher-than-expected incidence of HIV infections.
ASPIRE study
In addition to the overall 27% reduction in infections seen in ASPIRE, the investigators found that when they excluded data from two sites with lower rates of retention and adherence, the ring reduced infections by 37%.
A post hoc analysis showed significantly better protection for women over age 21 (P less than .001), but not for younger women (P = .45), a difference that correlated with reduced adherence.
The researchers noted that among women who acquired HIV, the rates of adverse medical events and resistance to antiretroviral agents were similar between the active ring and placebo groups.
Ring study
As of Oct. 16, 2015 – the data cutoff point for RING – there were 2,805 person-years of follow-up, and 761 women had completed 2 years of follow-up. There were a total of 133 post-randomization HIV-1 infections, 77 of which occurred among women assigned to the dapivirine ring, translating into an incidence of 4.08 per 100 person-years, and 56 of which occurred among women assigned to placebo, for an incidence of 6.10 per 100 person-years.
The overall reduction in infections in the active ring group in the Ring study relative to placebo was 31% (P = .0401). In a subgroup analysis of women over age 21, the dapivirine ring reduced infections by 37%.
The most common ring-related adverse events in this trial were abnormal uterine bleeding, pelvic discomfort and/or pain, suprapubic pain, and application site pain. Adverse-event rates, including product-related events, urogenital events, serious adverse events and death, were similar between the treatment arms.
Based on the results of the two trials, the International Partnership for Microbicides is planning an open-label extension study for the Ring cohort and submission of data for regulatory approval of the device.
The ASPIRE study was funded by the National Institutes of Health. The Ring Study was funded by the Bill and Melinda Gates Foundation, the U.S. President’s Emergency Plan for AIDS Relief, the U.S. Agency for International Development, and several European governments and organizations, and was conducted by the International Partnership for Microbicides.
Dr. Baeten and Dr. Nel reported having no financial disclosures.
AT CROI 2016
<p><b>Key clinical point:</b>An antiretroviral vaginal ring was modestly effective at reducing HIV infections versus placebo in high-risk women.
</p><p><b>Major finding:</b> The dapivirine ring reduced HIV infections by 27% in the ASPIRE trial, and by 31% in the Ring Study.
</p><p><b>Data source:</b> Phase III randomized trials conducted in Malawi, South Africa, Uganda, and Zimbabwe.
</p><p><b>Disclosures: </b>The ASPIRE study was funded by the National Institutes of Health. The Ring Study was funded by the Bill and Melinda Gates Foundation, the U.S. President’s Emergency Plan for AIDS Relief, the U.S. Agency for International Development, and several European governments and organizations, and was conducted by the International Partnership for Microbicides. Dr. Baeten and Dr. Nel reported having no financial disclosures.</p>

Smoking after breast cancer diagnosis a risk factor in cancer death
Women who smoke before or after a diagnosis of breast cancer have a significantly higher risk for death from breast cancer, respiratory tract cancers, and other causes than never smokers or quitters, follow-up results of a population-based prospective observation study show.
Among a subcohort of 4,562 women from the ages of 20 to 70, those who were active smokers within 1 year of a breast cancer diagnosis had a 25% greater risk for death from breast cancer, 14-fold higher risk for death from respiratory cancer, 6-fold risk for death from other respiratory diseases, and 2-fold higher risk for death from cardiovascular disease, found Dr. Michael N. Passarelli of the University of California, San Francisco, and his colleagues.

“Our study reinforces the importance of cigarette smoking cessation in women with breast cancer. For the minority of breast cancer survivors who continue to smoke after their diagnoses, these results should provide additional motivation to quit,” they write (J Clin Oncol. 2016 Jan 25. doi: 10.1200/JCO.2015.63.9328).
The investigators studied a cohort of 4,562 women who had taken part in the Collaborative Breast Cancer and Women’s Longevity Study, conducted in Massachusetts, New Hampshire, and Wisconsin. The study enrolled 20,691 women diagnosed from 1988 through 2008 with incident localized or regional invasive breast cancer.
The investigators re-contacted 4,562 participants a median of 6 years after their diagnosis. For women who reported smoking after breast cancer diagnosis, they calculated survival from the date of return of the questionnaire to the date of death or the end of follow-up.
The authors also created pre- and post-diagnosis proportional hazard regression models controlling for body mass index, education, parous status, age at first birth, menopausal status, family history of breast cancer, use of post-menopausal hormones, alcohol consumption, and the number of years between date of diagnosis and return of the study questionnaire.
For women who reported being active smokers within 1 year before a breast cancer diagnosis, hazard ratios (HR) for death from various causes were as follows (all statistically significant as shown by confidence intervals): breast cancer, HR 1.25; respiratory cancer, HR 14.48; other respiratory disease, HR 6.02; cardiovascular disease, HR 2.08.
For the 434 women (10%) who reported active smoking after diagnosis, the HR for breast-cancer death vs. never smokers was 1.72. Compared with women who continued to smoke, women who quit smoking after diagnosis had a lower risk for both breast-cancer death (a non-significant trend) and respiratory-cancer deaths (HR 0.39).
Women who smoke before or after a diagnosis of breast cancer have a significantly higher risk for death from breast cancer, respiratory tract cancers, and other causes than never smokers or quitters, follow-up results of a population-based prospective observation study show.
Among a subcohort of 4,562 women from the ages of 20 to 70, those who were active smokers within 1 year of a breast cancer diagnosis had a 25% greater risk for death from breast cancer, 14-fold higher risk for death from respiratory cancer, 6-fold risk for death from other respiratory diseases, and 2-fold higher risk for death from cardiovascular disease, found Dr. Michael N. Passarelli of the University of California, San Francisco, and his colleagues.
“Our study reinforces the importance of cigarette smoking cessation in women with breast cancer. For the minority of breast cancer survivors who continue to smoke after their diagnoses, these results should provide additional motivation to quit,” they write (J Clin Oncol. 2016 Jan 25. doi: 10.1200/JCO.2015.63.9328).
The investigators studied a cohort of 4,562 women who had taken part in the Collaborative Breast Cancer and Women’s Longevity Study, conducted in Massachusetts, New Hampshire, and Wisconsin. The study enrolled 20,691 women diagnosed from 1988 through 2008 with incident localized or regional invasive breast cancer.
The investigators re-contacted 4,562 participants a median of 6 years after their diagnosis. For women who reported smoking after breast cancer diagnosis, they calculated survival from the date of return of the questionnaire to the date of death or the end of follow-up.
The authors also created pre- and post-diagnosis proportional hazard regression models controlling for body mass index, education, parous status, age at first birth, menopausal status, family history of breast cancer, use of post-menopausal hormones, alcohol consumption, and the number of years between date of diagnosis and return of the study questionnaire.
For women who reported being active smokers within 1 year before a breast cancer diagnosis, hazard ratios (HR) for death from various causes were as follows (all statistically significant as shown by confidence intervals): breast cancer, HR 1.25; respiratory cancer, HR 14.48; other respiratory disease, HR 6.02; cardiovascular disease, HR 2.08.
For the 434 women (10%) who reported active smoking after diagnosis, the HR for breast-cancer death vs. never smokers was 1.72. Compared with women who continued to smoke, women who quit smoking after diagnosis had a lower risk for both breast-cancer death (a non-significant trend) and respiratory-cancer deaths (HR 0.39).
Women who smoke before or after a diagnosis of breast cancer have a significantly higher risk for death from breast cancer, respiratory tract cancers, and other causes than never smokers or quitters, follow-up results of a population-based prospective observation study show.
Among a subcohort of 4,562 women from the ages of 20 to 70, those who were active smokers within 1 year of a breast cancer diagnosis had a 25% greater risk for death from breast cancer, 14-fold higher risk for death from respiratory cancer, 6-fold risk for death from other respiratory diseases, and 2-fold higher risk for death from cardiovascular disease, found Dr. Michael N. Passarelli of the University of California, San Francisco, and his colleagues.
“Our study reinforces the importance of cigarette smoking cessation in women with breast cancer. For the minority of breast cancer survivors who continue to smoke after their diagnoses, these results should provide additional motivation to quit,” they write (J Clin Oncol. 2016 Jan 25. doi: 10.1200/JCO.2015.63.9328).
The investigators studied a cohort of 4,562 women who had taken part in the Collaborative Breast Cancer and Women’s Longevity Study, conducted in Massachusetts, New Hampshire, and Wisconsin. The study enrolled 20,691 women diagnosed from 1988 through 2008 with incident localized or regional invasive breast cancer.
The investigators re-contacted 4,562 participants a median of 6 years after their diagnosis. For women who reported smoking after breast cancer diagnosis, they calculated survival from the date of return of the questionnaire to the date of death or the end of follow-up.
The authors also created pre- and post-diagnosis proportional hazard regression models controlling for body mass index, education, parous status, age at first birth, menopausal status, family history of breast cancer, use of post-menopausal hormones, alcohol consumption, and the number of years between date of diagnosis and return of the study questionnaire.
For women who reported being active smokers within 1 year before a breast cancer diagnosis, hazard ratios (HR) for death from various causes were as follows (all statistically significant as shown by confidence intervals): breast cancer, HR 1.25; respiratory cancer, HR 14.48; other respiratory disease, HR 6.02; cardiovascular disease, HR 2.08.
For the 434 women (10%) who reported active smoking after diagnosis, the HR for breast-cancer death vs. never smokers was 1.72. Compared with women who continued to smoke, women who quit smoking after diagnosis had a lower risk for both breast-cancer death (a non-significant trend) and respiratory-cancer deaths (HR 0.39).
FROM THE JOURNAL OF CLINICAL ONCOLOGY
<p><b>Key clinical point: </b>Smoking is a significant risk factor for death from breast cancer and other causes.<b></b>
</p><p><b>Major finding: </b>Women who actively smoked within a year before a breast cancer diagnosis had a 25% higher risk for breast cancer death than never smokers.
</p><p><b>Data source: </b>Follow-up cohort of 4,562 women from a population-based prospective cohort study.<b></b>
</p><p><b>Disclosures: </b>The study was funded by grants from the National Cancer Institute and Susan G. Komen Foundation. The authors had no relevant conflicts of interest to disclose.</p>

Adjuvant oxaliplatin reduces colon cancer recurrence, deaths
Adding oxaliplatin to fluoropyrimidine-based adjuvant chemotherapy significantly reduces the risk of recurrence of colon cancer, an analysis of pooled data published Jan. 25 from five modern adjuvant therapy trials suggests.
Among more than 12,000 patients enrolled in the randomized clinical trials, the addition of oxaliplatin to standard adjuvant chemotherapy with fluorouracil (FU) and leucovorin was associated with significant reductions in the risk of recurrence within the first 14 months following treatment in patients with stage II colon cancer, and within the first 4 years for patients with stage III disease, report Dr. Manish A. Shah of Weill Cornell Medical College and Presbyterian Hospital, New York, and his colleagues.

“[W]e found that oxaliplatin significantly reduces the risk of recurrence and death within the first 6 years post treatment, with the greatest benefit observed in patients with higher-risk cancers. The time course risk of recurrence for stage II colon cancer is significantly reduced compared with patients with stage III disease, with potential implications for surveillance strategies,” they wrote (J Clin Onc. 2016 Jan 25. doi: 10.1200/JCO.2015.63.0558).
The authors looked at pooled data from five randomized studies with oxaliplatin for which mature data are available: the NSABP C-07 (National Surgical Adjuvant Breast and Bowel Project) trial, the NSABP C-08, the N0147, MOSAIC (Multicenter International Study of Oxaliplatin/ 5FU-LV in the Adjuvant Treatment of Colon Cancer), and XELOXA (Adjuvant XELOX).
They plotted continuous-time hazards of recurrence and death from the time of randomization up to 6 years in all patients treated with oxaliplatin and in those randomized to FU/leucovorin with or without oxaliplatin.
They found that for patients treated with FU, leucovorin, and oxaliplatin, the maximum hazard for recurrence occurred at around 14 months post treatment, and then dwindled to nearly zero after about 6 years of follow-up. They also found that the risk of recurrence was lower with the addition of oxaliplatin at all-time points.
For the endpoint of death, oxaliplatin significantly reduced the risk from 2 to 6 years after treatment in patients with stage III disease. The addition of oxaliplatin, however, did not significantly reduce the risk of death for patients with stage II disease at any time point. The investigators also found that oxaliplatin was least effective at preventing recurrences in patients with stage II disease, suggesting a limited role for intensive adjuvant therapy in this group.
“These data support the hypothesis that the addition of oxaliplatin to fluoropyrimidine therapy provides sustained benefit over time, preventing recurrences that would ultimately lead to deaths in this large patient population,” the authors wrote.The analysis was supported by a grant from the National Cancer Institute. Dr. Shah disclosed consulting/advising Eli Lilly/ImClone Systems and Genentech, and institutional research funding from Sanofi and Genentech.
Adding oxaliplatin to fluoropyrimidine-based adjuvant chemotherapy significantly reduces the risk of recurrence of colon cancer, an analysis of pooled data published Jan. 25 from five modern adjuvant therapy trials suggests.
Among more than 12,000 patients enrolled in the randomized clinical trials, the addition of oxaliplatin to standard adjuvant chemotherapy with fluorouracil (FU) and leucovorin was associated with significant reductions in the risk of recurrence within the first 14 months following treatment in patients with stage II colon cancer, and within the first 4 years for patients with stage III disease, report Dr. Manish A. Shah of Weill Cornell Medical College and Presbyterian Hospital, New York, and his colleagues.
“[W]e found that oxaliplatin significantly reduces the risk of recurrence and death within the first 6 years post treatment, with the greatest benefit observed in patients with higher-risk cancers. The time course risk of recurrence for stage II colon cancer is significantly reduced compared with patients with stage III disease, with potential implications for surveillance strategies,” they wrote (J Clin Onc. 2016 Jan 25. doi: 10.1200/JCO.2015.63.0558).
The authors looked at pooled data from five randomized studies with oxaliplatin for which mature data are available: the NSABP C-07 (National Surgical Adjuvant Breast and Bowel Project) trial, the NSABP C-08, the N0147, MOSAIC (Multicenter International Study of Oxaliplatin/ 5FU-LV in the Adjuvant Treatment of Colon Cancer), and XELOXA (Adjuvant XELOX).
They plotted continuous-time hazards of recurrence and death from the time of randomization up to 6 years in all patients treated with oxaliplatin and in those randomized to FU/leucovorin with or without oxaliplatin.
They found that for patients treated with FU, leucovorin, and oxaliplatin, the maximum hazard for recurrence occurred at around 14 months post treatment, and then dwindled to nearly zero after about 6 years of follow-up. They also found that the risk of recurrence was lower with the addition of oxaliplatin at all-time points.
For the endpoint of death, oxaliplatin significantly reduced the risk from 2 to 6 years after treatment in patients with stage III disease. The addition of oxaliplatin, however, did not significantly reduce the risk of death for patients with stage II disease at any time point. The investigators also found that oxaliplatin was least effective at preventing recurrences in patients with stage II disease, suggesting a limited role for intensive adjuvant therapy in this group.
“These data support the hypothesis that the addition of oxaliplatin to fluoropyrimidine therapy provides sustained benefit over time, preventing recurrences that would ultimately lead to deaths in this large patient population,” the authors wrote.The analysis was supported by a grant from the National Cancer Institute. Dr. Shah disclosed consulting/advising Eli Lilly/ImClone Systems and Genentech, and institutional research funding from Sanofi and Genentech.
Adding oxaliplatin to fluoropyrimidine-based adjuvant chemotherapy significantly reduces the risk of recurrence of colon cancer, an analysis of pooled data published Jan. 25 from five modern adjuvant therapy trials suggests.
Among more than 12,000 patients enrolled in the randomized clinical trials, the addition of oxaliplatin to standard adjuvant chemotherapy with fluorouracil (FU) and leucovorin was associated with significant reductions in the risk of recurrence within the first 14 months following treatment in patients with stage II colon cancer, and within the first 4 years for patients with stage III disease, report Dr. Manish A. Shah of Weill Cornell Medical College and Presbyterian Hospital, New York, and his colleagues.
“[W]e found that oxaliplatin significantly reduces the risk of recurrence and death within the first 6 years post treatment, with the greatest benefit observed in patients with higher-risk cancers. The time course risk of recurrence for stage II colon cancer is significantly reduced compared with patients with stage III disease, with potential implications for surveillance strategies,” they wrote (J Clin Onc. 2016 Jan 25. doi: 10.1200/JCO.2015.63.0558).
The authors looked at pooled data from five randomized studies with oxaliplatin for which mature data are available: the NSABP C-07 (National Surgical Adjuvant Breast and Bowel Project) trial, the NSABP C-08, the N0147, MOSAIC (Multicenter International Study of Oxaliplatin/ 5FU-LV in the Adjuvant Treatment of Colon Cancer), and XELOXA (Adjuvant XELOX).
They plotted continuous-time hazards of recurrence and death from the time of randomization up to 6 years in all patients treated with oxaliplatin and in those randomized to FU/leucovorin with or without oxaliplatin.
They found that for patients treated with FU, leucovorin, and oxaliplatin, the maximum hazard for recurrence occurred at around 14 months post treatment, and then dwindled to nearly zero after about 6 years of follow-up. They also found that the risk of recurrence was lower with the addition of oxaliplatin at all-time points.
For the endpoint of death, oxaliplatin significantly reduced the risk from 2 to 6 years after treatment in patients with stage III disease. The addition of oxaliplatin, however, did not significantly reduce the risk of death for patients with stage II disease at any time point. The investigators also found that oxaliplatin was least effective at preventing recurrences in patients with stage II disease, suggesting a limited role for intensive adjuvant therapy in this group.
“These data support the hypothesis that the addition of oxaliplatin to fluoropyrimidine therapy provides sustained benefit over time, preventing recurrences that would ultimately lead to deaths in this large patient population,” the authors wrote.The analysis was supported by a grant from the National Cancer Institute. Dr. Shah disclosed consulting/advising Eli Lilly/ImClone Systems and Genentech, and institutional research funding from Sanofi and Genentech.
<p><b>Key clinical point:</b> Adding oxaliplatin to fluoropyrimidine-based adjuvant chemotherapy significantly reduces the risk of recurrence of colon cancer, especially stage III.
</p><p><b>Major finding:</b> The addition of oxaliplatin was associated with lower risk of recurrence within the first 14 months in stage II and within the first 4 months in stage III disease.
</p><p><b>Data source:</b> Pooled analysis of data from five randomized clinical trials.
</p><p><b>Disclosures:</b> The analysis was supported by a grant from the National Cancer Institute. Dr. Shah disclosed consulting/advising Eli Lilly/ImClone Systems and Genentech, and institutional research funding from Sanofi and Genentech.</p>

PROCLAIM: Pemetrexed combo no better than standard tx in NSCLC
A combination of the antifolate agent pemetrexed plus cisplatin with thoracic radiation followed by consolidation with pemetrexed was no better than standard etoposide-cisplatin chemoradiotherapy and platinum doublet consolidation in patients with unresectable stage III nonsquamous, non–small cell lung cancer, results of a randomized clinical trial indicate.
In the PROCLAIM trial, results of which were reported at the American Society of Clinical Oncology’s 2015 annual meeting, there was no significant difference in overall survival between patients randomized to receive pemetrexed-cisplatin or etoposide-cisplatin, leading to early discontinuation of the trial because it had met the prespecified definition of futility, reported Dr. Suresh Senan from the Vrije University Medical Center in Amsterdam and colleagues.
They noted, however, that pemetrexed-cisplatin was less toxic than the current platinum doublet standard.
“A significantly lower incidence of drug-related grade 3 to 4 [adverse events], including neutropenia, was observed during the overall study period for pemetrexed-cisplatin. Grade 3 to 4 neutropenia and febrile neutropenia were also lower for pemetrexed-cisplatin during concurrent therapy. The PROCLAIM trial showed that pemetrexed-cisplatin has an acceptable safety profile, in a scenario in which concurrent chemoradiation remains the standard of care,” the investigators reported in the Journal of Clinical Oncology.
Investigators in the multicenter international trial chose to compare pemetrexed in combination with cisplatin because the antifolate has selective activity against nonsquamous NSCLC and is known to be a radiosensitizer. Additionally, maintenance pemetrexed has been shown in at least two randomized trials to offer a survival advantage following first-line therapy, they noted (J Clin Onc. 2016 Jan 25. doi: 10.1200/JCO.2015.64.8824).
In the PROCLAIM trial, patients with stage IIIA or IIIB unresectable nonsquamous NSCLC were randomly assigned to receive either pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 intravenously every 3 weeks for three cycles with concurrent thoracic radiation at doses of 60 to 66 Gy, followed by pemetrexed consolidation every 3 weeks for four cycles, or to standard therapy with etoposide 50 mg/m2 and cisplatin 50 mg/m2 intravenously every 4 weeks for two cycles with concurrent thoracic radiation at the same doses, followed by two cycles of consolidation with platinum-based doublet chemotherapy.
The trial was designed as a superiority study, with planned enrollment of about 600 patients, with 80% power to detect a hazard ratio of 0.74 for the experimental arm. The trial was stopped early for futility after an interim analysis was performed. At that time, 173 of 552 randomized patients had died.
Median overall survival was 26.8 months for the pemetrexed group vs. 25 months for the standard therapy group (HR, 0.98; P = .831).
A combination of the antifolate agent pemetrexed plus cisplatin with thoracic radiation followed by consolidation with pemetrexed was no better than standard etoposide-cisplatin chemoradiotherapy and platinum doublet consolidation in patients with unresectable stage III nonsquamous, non–small cell lung cancer, results of a randomized clinical trial indicate.
In the PROCLAIM trial, results of which were reported at the American Society of Clinical Oncology’s 2015 annual meeting, there was no significant difference in overall survival between patients randomized to receive pemetrexed-cisplatin or etoposide-cisplatin, leading to early discontinuation of the trial because it had met the prespecified definition of futility, reported Dr. Suresh Senan from the Vrije University Medical Center in Amsterdam and colleagues.
They noted, however, that pemetrexed-cisplatin was less toxic than the current platinum doublet standard.
“A significantly lower incidence of drug-related grade 3 to 4 [adverse events], including neutropenia, was observed during the overall study period for pemetrexed-cisplatin. Grade 3 to 4 neutropenia and febrile neutropenia were also lower for pemetrexed-cisplatin during concurrent therapy. The PROCLAIM trial showed that pemetrexed-cisplatin has an acceptable safety profile, in a scenario in which concurrent chemoradiation remains the standard of care,” the investigators reported in the Journal of Clinical Oncology.
Investigators in the multicenter international trial chose to compare pemetrexed in combination with cisplatin because the antifolate has selective activity against nonsquamous NSCLC and is known to be a radiosensitizer. Additionally, maintenance pemetrexed has been shown in at least two randomized trials to offer a survival advantage following first-line therapy, they noted (J Clin Onc. 2016 Jan 25. doi: 10.1200/JCO.2015.64.8824).
In the PROCLAIM trial, patients with stage IIIA or IIIB unresectable nonsquamous NSCLC were randomly assigned to receive either pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 intravenously every 3 weeks for three cycles with concurrent thoracic radiation at doses of 60 to 66 Gy, followed by pemetrexed consolidation every 3 weeks for four cycles, or to standard therapy with etoposide 50 mg/m2 and cisplatin 50 mg/m2 intravenously every 4 weeks for two cycles with concurrent thoracic radiation at the same doses, followed by two cycles of consolidation with platinum-based doublet chemotherapy.
The trial was designed as a superiority study, with planned enrollment of about 600 patients, with 80% power to detect a hazard ratio of 0.74 for the experimental arm. The trial was stopped early for futility after an interim analysis was performed. At that time, 173 of 552 randomized patients had died.
Median overall survival was 26.8 months for the pemetrexed group vs. 25 months for the standard therapy group (HR, 0.98; P = .831).
A combination of the antifolate agent pemetrexed plus cisplatin with thoracic radiation followed by consolidation with pemetrexed was no better than standard etoposide-cisplatin chemoradiotherapy and platinum doublet consolidation in patients with unresectable stage III nonsquamous, non–small cell lung cancer, results of a randomized clinical trial indicate.
In the PROCLAIM trial, results of which were reported at the American Society of Clinical Oncology’s 2015 annual meeting, there was no significant difference in overall survival between patients randomized to receive pemetrexed-cisplatin or etoposide-cisplatin, leading to early discontinuation of the trial because it had met the prespecified definition of futility, reported Dr. Suresh Senan from the Vrije University Medical Center in Amsterdam and colleagues.
They noted, however, that pemetrexed-cisplatin was less toxic than the current platinum doublet standard.
“A significantly lower incidence of drug-related grade 3 to 4 [adverse events], including neutropenia, was observed during the overall study period for pemetrexed-cisplatin. Grade 3 to 4 neutropenia and febrile neutropenia were also lower for pemetrexed-cisplatin during concurrent therapy. The PROCLAIM trial showed that pemetrexed-cisplatin has an acceptable safety profile, in a scenario in which concurrent chemoradiation remains the standard of care,” the investigators reported in the Journal of Clinical Oncology.
Investigators in the multicenter international trial chose to compare pemetrexed in combination with cisplatin because the antifolate has selective activity against nonsquamous NSCLC and is known to be a radiosensitizer. Additionally, maintenance pemetrexed has been shown in at least two randomized trials to offer a survival advantage following first-line therapy, they noted (J Clin Onc. 2016 Jan 25. doi: 10.1200/JCO.2015.64.8824).
In the PROCLAIM trial, patients with stage IIIA or IIIB unresectable nonsquamous NSCLC were randomly assigned to receive either pemetrexed 500 mg/m2 and cisplatin 75 mg/m2 intravenously every 3 weeks for three cycles with concurrent thoracic radiation at doses of 60 to 66 Gy, followed by pemetrexed consolidation every 3 weeks for four cycles, or to standard therapy with etoposide 50 mg/m2 and cisplatin 50 mg/m2 intravenously every 4 weeks for two cycles with concurrent thoracic radiation at the same doses, followed by two cycles of consolidation with platinum-based doublet chemotherapy.
The trial was designed as a superiority study, with planned enrollment of about 600 patients, with 80% power to detect a hazard ratio of 0.74 for the experimental arm. The trial was stopped early for futility after an interim analysis was performed. At that time, 173 of 552 randomized patients had died.
Median overall survival was 26.8 months for the pemetrexed group vs. 25 months for the standard therapy group (HR, 0.98; P = .831).
FROM THE JOURNAL OF CLINICAL ONCOLOGY
<p><b>Key clinical point:</b> Pemetrexed-cisplatin and pemetrexed consolidation were not superior to etoposide-cisplatin and consolidation for advanced NSCLC.
</p><p><b>Major finding:</b> Median OS was 26.8 months for the pemetrexed group vs. 25 for the standard therapy group (HR, 0.98; <i>P</i> = .831).
</p><p><b>Data source:</b> Randomized phase III trial planned for 598 patients with advanced nonsquamous, non–small cell lung cancer.
</p><p><b>Disclosures:</b> The study was supported by Eli Lilly. Multiple authors reported research funding, consulting, or honoraria from the company, and six are Eli Lilly employees.</p>
MAPK inhibitor combo offers prolonged survival from BRAF+ advanced melanoma
Combination therapy to inhibit the MAP kinase pathway is associated with a median overall survival of more than 2 years for patients with metastatic melanoma positive for the BRAF V600 mutation who have not previously received a BRAF inhibitor.
An analysis of data from a phase I and II trial of the BRAF inhibitor dabrafenib (Tafinlar), 150 mg twice daily, and the MEK inhibitor trametinib (Mekinist), 2 mg once daily, in 78 BRAF inhibitor–naïve patients with metastatic melanoma showed that in a non-randomized cohort (part B) the median overall survival (OS) was 27.4 months. In a cohort randomized to the same doses (part C), the median OS was 25 months, reported Dr. Georgina V. Long and her colleagues from the Melanoma Institute Australia and the University of Sydney in the Journal of Clinical Oncology.
Among 24 patients treated in part B, progression-free survival (PFS) was 18% at 3 years.
“The combination has an acceptable long-term safety profile and is a standard of care for patients with BRAF mutation–positive metastatic melanoma, particularly given the recent publications demonstrating a significant improvement in the PFS and OS in phase III trials of combination versus single-agent BRAF inhibitors,” the investigators wrote (J Clin Oncol. 2016 Jan 25. doi: 10.1200/JCO.2015.62.9345).
The investigators conducted the analysis to determine which patients were the most likely to benefit from combination therapy of the MAP (mitogen-active protein) kinase pathway.
They observed that in the part B, non-randomized cohort, OS was 72% at 1 year, 60% at 2 tears, and 47% at 3 years. In the part C, randomized cohort, OS was 80%, 51%, and 38%, respectively.
Factors associated with better OS in Cox proportional hazards regression models included metastases to fewer than three organs sites, and lower levels of lactate dehydrogenase (LDH) at baseline. Not surprisingly, a complete response to therapy was also associated with better survival. Three-year OS rates were 62% for patients with normal baseline LDH levels, and 63% for patients who had a complete response.
Combination therapy to inhibit the MAP kinase pathway is associated with a median overall survival of more than 2 years for patients with metastatic melanoma positive for the BRAF V600 mutation who have not previously received a BRAF inhibitor.
An analysis of data from a phase I and II trial of the BRAF inhibitor dabrafenib (Tafinlar), 150 mg twice daily, and the MEK inhibitor trametinib (Mekinist), 2 mg once daily, in 78 BRAF inhibitor–naïve patients with metastatic melanoma showed that in a non-randomized cohort (part B) the median overall survival (OS) was 27.4 months. In a cohort randomized to the same doses (part C), the median OS was 25 months, reported Dr. Georgina V. Long and her colleagues from the Melanoma Institute Australia and the University of Sydney in the Journal of Clinical Oncology.
Among 24 patients treated in part B, progression-free survival (PFS) was 18% at 3 years.
“The combination has an acceptable long-term safety profile and is a standard of care for patients with BRAF mutation–positive metastatic melanoma, particularly given the recent publications demonstrating a significant improvement in the PFS and OS in phase III trials of combination versus single-agent BRAF inhibitors,” the investigators wrote (J Clin Oncol. 2016 Jan 25. doi: 10.1200/JCO.2015.62.9345).
The investigators conducted the analysis to determine which patients were the most likely to benefit from combination therapy of the MAP (mitogen-active protein) kinase pathway.
They observed that in the part B, non-randomized cohort, OS was 72% at 1 year, 60% at 2 tears, and 47% at 3 years. In the part C, randomized cohort, OS was 80%, 51%, and 38%, respectively.
Factors associated with better OS in Cox proportional hazards regression models included metastases to fewer than three organs sites, and lower levels of lactate dehydrogenase (LDH) at baseline. Not surprisingly, a complete response to therapy was also associated with better survival. Three-year OS rates were 62% for patients with normal baseline LDH levels, and 63% for patients who had a complete response.
Combination therapy to inhibit the MAP kinase pathway is associated with a median overall survival of more than 2 years for patients with metastatic melanoma positive for the BRAF V600 mutation who have not previously received a BRAF inhibitor.
An analysis of data from a phase I and II trial of the BRAF inhibitor dabrafenib (Tafinlar), 150 mg twice daily, and the MEK inhibitor trametinib (Mekinist), 2 mg once daily, in 78 BRAF inhibitor–naïve patients with metastatic melanoma showed that in a non-randomized cohort (part B) the median overall survival (OS) was 27.4 months. In a cohort randomized to the same doses (part C), the median OS was 25 months, reported Dr. Georgina V. Long and her colleagues from the Melanoma Institute Australia and the University of Sydney in the Journal of Clinical Oncology.
Among 24 patients treated in part B, progression-free survival (PFS) was 18% at 3 years.
“The combination has an acceptable long-term safety profile and is a standard of care for patients with BRAF mutation–positive metastatic melanoma, particularly given the recent publications demonstrating a significant improvement in the PFS and OS in phase III trials of combination versus single-agent BRAF inhibitors,” the investigators wrote (J Clin Oncol. 2016 Jan 25. doi: 10.1200/JCO.2015.62.9345).
The investigators conducted the analysis to determine which patients were the most likely to benefit from combination therapy of the MAP (mitogen-active protein) kinase pathway.
They observed that in the part B, non-randomized cohort, OS was 72% at 1 year, 60% at 2 tears, and 47% at 3 years. In the part C, randomized cohort, OS was 80%, 51%, and 38%, respectively.
Factors associated with better OS in Cox proportional hazards regression models included metastases to fewer than three organs sites, and lower levels of lactate dehydrogenase (LDH) at baseline. Not surprisingly, a complete response to therapy was also associated with better survival. Three-year OS rates were 62% for patients with normal baseline LDH levels, and 63% for patients who had a complete response.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
<p><b>Key clinical point: </b>Combination MAP kinase pathway inhibition was associated with prolonged survival of BRAF mutation-positive metastatic melanoma.
</p><p><b>Major finding: </b>Median overall survival was 27.4 months in a non-randomized cohort, and 25 months in a randomized cohort.
</p><p><b>Data source: </b>Analysis of a subset of patients in a phase I and II trial of dabrafenib and trametinib.
</p><p><b>Disclosures: </b>The study was supported by GlaxoSmithKline. Dr. Long and many coauthors disclosed consulting and/or research funding from the company.</p>
European societies issue aspergillosis diagnosis, management guidelines
European respiratory disease and infectious disease specialists have banded together to issue new clinical guidelines on the diagnosis and management of an uncommon but serious problem: chronic pulmonary aspergillosis (CPA).
Pulmonary infections with Aspergillus species, although uncommon, are a complicating factor in several lung diseases, especially tuberculosis, and aspergillosis is a serious, often fatal opportunistic infection in transplant recipients who are on chronic immunosuppression or patients who are immunocompromised from disease or cytotoxic chemotherapy.
Approximately 240,000 people in Europe and 3 million people worldwide have chronic pulmonary aspergillosis (CPA). The Centers for Disease Control and Prevention notes that because aspergillosis is not classified as a reportable disease, data on the actual incidence of infections in the United States are hard to come by.
“You don’t see this every day, whether you’re an infectious disease specialist or pulmonologist, so you really can’t rely on your experience to guide you in managing these cases, which is why guidelines such as this can be very helpful,” commented Dr. Norman Edelman, a pulmonologist and senior consultant for scientific affairs for the American Lung Association.
The guidelines, issued by the European Society for Clinical Microbiology and Infectious Diseases in cooperation with the European Confederation of Medical Mycology and the European Respiratory Society, are an attempt to provide clinicians with the best possible evidence-based guidance on managing patients with aspergillosis, primarily those with CPA (Eur Respir J. 2015. doi: 10.1183/13993003.00583-2015).
Dr. Edelman noted that the most frequent presentation he sees – and that very infrequently – is allergic bronchopulmonary aspergillosis in patients with asthma.
The most recent U.S. guidelines, issued under the aegis of the Infectious Diseases Society of America (IDSA) in 2000 and revised in 2008 (CID 2008;46:327-360), differ from the European recommendations in their level of detail, explained Prof. David W. Denning, professor of infectious diseases in global health at the University of Manchester (England) and lead author of the European guidelines.
“The IDSA guidelines assume that you know how to make the diagnosis, but actually for chronic pulmonary aspergillosis that’s not so easy with some patients,” he said in an interview.
“The European ones go into in great detail the diagnosis, the radiology, whether this test is better than that test, how they all add up, and all that sort of stuff,” he said,
The European guidelines also make recommendations for duration of therapy and comment on the use of steroids and immunotherapy with interferon-gamma, Dr. Denning noted.
Diagnostic criteria
The European guidelines categorize Aspergillus infections according to differences in clinical management:
• Simple aspergilloma. A single pulmonary cavity containing a fungal ball, supported by serologic or microbiologic evidence of infections with Aspergillus species in patients who are not immunocompromised and are asymptomatic or have only minor symptoms and no radiographic evidence of progression for at least 3 months.
• Chronic cavitary pulmonary aspergillosis (CCPA). The presence of one or more pulmonary cavities that may contain one or more aspergillomas or irregular intraluminal material, evidence of Aspergillus species, significant pulmonary/systemic symptoms, and overt progression on radiography over 3 or more months of observations.
• Chronic fibrosing pulmonary aspergillosis (CFPA). Severe, fibrotic destruction of at least two lung lobes as a complication of CCPA, causing a major loss of lung function. The guidelines note that destruction of a single lobe is designated as CCPA of that lobe.
• Aspergillus nodules. This unusual presentation is marked by the presence of one or more nodules that may or may not cavitate. The nodules may resemble tuberculoma, carcinoma of the lung, or coccidioidomycosis; histology is required to make an accurate diagnosis.
• Subacute invasive aspergillosis (SAIA). This can occur over the course of 1-3 months in patients who are mildly immunocompromised. Radiologic features can vary, and may include cavitation, the presence of nodules, and progressive consolidation with the appearance of abscess formation. Fungal hyphae (filaments) can be seen in biopsied lung tissues, and there may be evidence of Aspergillus galactomannan antigen in respiratory fluids or blood.
Treatment
The guidelines note that most of the evidence for managing CPA are based on cohort studies and case reports rather than randomized clinical trials, and that there have been no head-to-head trials comparing oral triazole agents.
For treatment of CPA, the European guidelines recommend:
• Itraconazole 200 mg twice daily, with therapeutic drug monitoring and dose adjustment as necessary (Grade A [strong] recommendation).
• Voriconazole 150-200 mg twice daily, with monitoring and dose adjustment. The guidelines recommend lower doses for patients older than 70 years, those with low body weight, significant liver disease, and/or those of Northeast Asian descent, who may be genetically inclined to slow drug metabolism (Grade A).
• Posaconazole liquid 400 mg twice daily, or tablets 300 mg once daily (Grade B [moderate] recommendation].
In general, the recommended duration of therapy for control of infection in patients with CPA or curative intent for patients with SAIA or chronic necrotizing pulmonary aspergillosis is 6 months or more, depending on patient status and drug tolerance.
For patients with CPA with progressive disease, those whom therapy has failed, or those who are intolerant of or have disease resistant to triazoles, intravenous therapy with micafungin, 150 mg day (Grade B); amphotericin B deoxycholate, 0.7-1.0 mg/kg per day (Grade C [marginal] recommendation); liposomal amphotericin B, 3 mg/kg per day (Grade B); or caspofungin, 50-70 mg/day (Grade C) are recommended.
The guidelines also recommend surgical excision of simple aspergilloma, preferably by a video-assisted thoracic surgery technique, if technically feasible.
“In my own experience, we resort to surgery very infrequently,” Dr. Edelman said.
He noted that it would be helpful if the guidelines had also allergic bronchopulmonary aspergillosis as a separate entity.
Ideal not always achievable
Prof. Denning points out that the optimum therapies and practices described in the guidelines can’t always be implemented. Worldwide, he said, antifungal therapy is not widely available, with the exception of fluconazole, which has no activity against Aspergillus, and is inferior to itraconazole and other extended azoles for other fungal diseases such as histoplasmosis, blastomycosis, and paracoccidioidomycosis.
The price of antifungal therapies can also be a barrier to effective treatment in many parts of the world.
“If you’re having to pay for your medicines and you’re living on $5 or $10 a day in Kenya, say, you can’t afford to buy them. So even if the drugs are physically there, it may not be really affordable for a course of therapy for these patients, and there’s some advocacy to be done around that for the whole world,” he said.
The guidelines were funded primarily by grants from ESCMID and ERS with additional support from ECMM. Authors’ travel expenses were funded jointly by ESCMID and ERS. Dr. Denning has received grant support and founder shares in F2G, and has received grants from the Fungal Research Trust, Wellcome Trust, Moulton Trust, Medical Research Council, Chronic Granulomatous Disease Research Trust, National Institute of Allergy and Infectious Diseases, National Institute of Health Research and the European Union, and AstraZeneca. Dr. Edelman reported no relevant disclosures.
European respiratory disease and infectious disease specialists have banded together to issue new clinical guidelines on the diagnosis and management of an uncommon but serious problem: chronic pulmonary aspergillosis (CPA).
Pulmonary infections with Aspergillus species, although uncommon, are a complicating factor in several lung diseases, especially tuberculosis, and aspergillosis is a serious, often fatal opportunistic infection in transplant recipients who are on chronic immunosuppression or patients who are immunocompromised from disease or cytotoxic chemotherapy.
Approximately 240,000 people in Europe and 3 million people worldwide have chronic pulmonary aspergillosis (CPA). The Centers for Disease Control and Prevention notes that because aspergillosis is not classified as a reportable disease, data on the actual incidence of infections in the United States are hard to come by.
“You don’t see this every day, whether you’re an infectious disease specialist or pulmonologist, so you really can’t rely on your experience to guide you in managing these cases, which is why guidelines such as this can be very helpful,” commented Dr. Norman Edelman, a pulmonologist and senior consultant for scientific affairs for the American Lung Association.
The guidelines, issued by the European Society for Clinical Microbiology and Infectious Diseases in cooperation with the European Confederation of Medical Mycology and the European Respiratory Society, are an attempt to provide clinicians with the best possible evidence-based guidance on managing patients with aspergillosis, primarily those with CPA (Eur Respir J. 2015. doi: 10.1183/13993003.00583-2015).
Dr. Edelman noted that the most frequent presentation he sees – and that very infrequently – is allergic bronchopulmonary aspergillosis in patients with asthma.
The most recent U.S. guidelines, issued under the aegis of the Infectious Diseases Society of America (IDSA) in 2000 and revised in 2008 (CID 2008;46:327-360), differ from the European recommendations in their level of detail, explained Prof. David W. Denning, professor of infectious diseases in global health at the University of Manchester (England) and lead author of the European guidelines.
“The IDSA guidelines assume that you know how to make the diagnosis, but actually for chronic pulmonary aspergillosis that’s not so easy with some patients,” he said in an interview.
“The European ones go into in great detail the diagnosis, the radiology, whether this test is better than that test, how they all add up, and all that sort of stuff,” he said,
The European guidelines also make recommendations for duration of therapy and comment on the use of steroids and immunotherapy with interferon-gamma, Dr. Denning noted.
Diagnostic criteria
The European guidelines categorize Aspergillus infections according to differences in clinical management:
• Simple aspergilloma. A single pulmonary cavity containing a fungal ball, supported by serologic or microbiologic evidence of infections with Aspergillus species in patients who are not immunocompromised and are asymptomatic or have only minor symptoms and no radiographic evidence of progression for at least 3 months.
• Chronic cavitary pulmonary aspergillosis (CCPA). The presence of one or more pulmonary cavities that may contain one or more aspergillomas or irregular intraluminal material, evidence of Aspergillus species, significant pulmonary/systemic symptoms, and overt progression on radiography over 3 or more months of observations.
• Chronic fibrosing pulmonary aspergillosis (CFPA). Severe, fibrotic destruction of at least two lung lobes as a complication of CCPA, causing a major loss of lung function. The guidelines note that destruction of a single lobe is designated as CCPA of that lobe.
• Aspergillus nodules. This unusual presentation is marked by the presence of one or more nodules that may or may not cavitate. The nodules may resemble tuberculoma, carcinoma of the lung, or coccidioidomycosis; histology is required to make an accurate diagnosis.
• Subacute invasive aspergillosis (SAIA). This can occur over the course of 1-3 months in patients who are mildly immunocompromised. Radiologic features can vary, and may include cavitation, the presence of nodules, and progressive consolidation with the appearance of abscess formation. Fungal hyphae (filaments) can be seen in biopsied lung tissues, and there may be evidence of Aspergillus galactomannan antigen in respiratory fluids or blood.
Treatment
The guidelines note that most of the evidence for managing CPA are based on cohort studies and case reports rather than randomized clinical trials, and that there have been no head-to-head trials comparing oral triazole agents.
For treatment of CPA, the European guidelines recommend:
• Itraconazole 200 mg twice daily, with therapeutic drug monitoring and dose adjustment as necessary (Grade A [strong] recommendation).
• Voriconazole 150-200 mg twice daily, with monitoring and dose adjustment. The guidelines recommend lower doses for patients older than 70 years, those with low body weight, significant liver disease, and/or those of Northeast Asian descent, who may be genetically inclined to slow drug metabolism (Grade A).
• Posaconazole liquid 400 mg twice daily, or tablets 300 mg once daily (Grade B [moderate] recommendation].
In general, the recommended duration of therapy for control of infection in patients with CPA or curative intent for patients with SAIA or chronic necrotizing pulmonary aspergillosis is 6 months or more, depending on patient status and drug tolerance.
For patients with CPA with progressive disease, those whom therapy has failed, or those who are intolerant of or have disease resistant to triazoles, intravenous therapy with micafungin, 150 mg day (Grade B); amphotericin B deoxycholate, 0.7-1.0 mg/kg per day (Grade C [marginal] recommendation); liposomal amphotericin B, 3 mg/kg per day (Grade B); or caspofungin, 50-70 mg/day (Grade C) are recommended.
The guidelines also recommend surgical excision of simple aspergilloma, preferably by a video-assisted thoracic surgery technique, if technically feasible.
“In my own experience, we resort to surgery very infrequently,” Dr. Edelman said.
He noted that it would be helpful if the guidelines had also allergic bronchopulmonary aspergillosis as a separate entity.
Ideal not always achievable
Prof. Denning points out that the optimum therapies and practices described in the guidelines can’t always be implemented. Worldwide, he said, antifungal therapy is not widely available, with the exception of fluconazole, which has no activity against Aspergillus, and is inferior to itraconazole and other extended azoles for other fungal diseases such as histoplasmosis, blastomycosis, and paracoccidioidomycosis.
The price of antifungal therapies can also be a barrier to effective treatment in many parts of the world.
“If you’re having to pay for your medicines and you’re living on $5 or $10 a day in Kenya, say, you can’t afford to buy them. So even if the drugs are physically there, it may not be really affordable for a course of therapy for these patients, and there’s some advocacy to be done around that for the whole world,” he said.
The guidelines were funded primarily by grants from ESCMID and ERS with additional support from ECMM. Authors’ travel expenses were funded jointly by ESCMID and ERS. Dr. Denning has received grant support and founder shares in F2G, and has received grants from the Fungal Research Trust, Wellcome Trust, Moulton Trust, Medical Research Council, Chronic Granulomatous Disease Research Trust, National Institute of Allergy and Infectious Diseases, National Institute of Health Research and the European Union, and AstraZeneca. Dr. Edelman reported no relevant disclosures.
European respiratory disease and infectious disease specialists have banded together to issue new clinical guidelines on the diagnosis and management of an uncommon but serious problem: chronic pulmonary aspergillosis (CPA).
Pulmonary infections with Aspergillus species, although uncommon, are a complicating factor in several lung diseases, especially tuberculosis, and aspergillosis is a serious, often fatal opportunistic infection in transplant recipients who are on chronic immunosuppression or patients who are immunocompromised from disease or cytotoxic chemotherapy.
Approximately 240,000 people in Europe and 3 million people worldwide have chronic pulmonary aspergillosis (CPA). The Centers for Disease Control and Prevention notes that because aspergillosis is not classified as a reportable disease, data on the actual incidence of infections in the United States are hard to come by.
“You don’t see this every day, whether you’re an infectious disease specialist or pulmonologist, so you really can’t rely on your experience to guide you in managing these cases, which is why guidelines such as this can be very helpful,” commented Dr. Norman Edelman, a pulmonologist and senior consultant for scientific affairs for the American Lung Association.
The guidelines, issued by the European Society for Clinical Microbiology and Infectious Diseases in cooperation with the European Confederation of Medical Mycology and the European Respiratory Society, are an attempt to provide clinicians with the best possible evidence-based guidance on managing patients with aspergillosis, primarily those with CPA (Eur Respir J. 2015. doi: 10.1183/13993003.00583-2015).
Dr. Edelman noted that the most frequent presentation he sees – and that very infrequently – is allergic bronchopulmonary aspergillosis in patients with asthma.
The most recent U.S. guidelines, issued under the aegis of the Infectious Diseases Society of America (IDSA) in 2000 and revised in 2008 (CID 2008;46:327-360), differ from the European recommendations in their level of detail, explained Prof. David W. Denning, professor of infectious diseases in global health at the University of Manchester (England) and lead author of the European guidelines.
“The IDSA guidelines assume that you know how to make the diagnosis, but actually for chronic pulmonary aspergillosis that’s not so easy with some patients,” he said in an interview.
“The European ones go into in great detail the diagnosis, the radiology, whether this test is better than that test, how they all add up, and all that sort of stuff,” he said,
The European guidelines also make recommendations for duration of therapy and comment on the use of steroids and immunotherapy with interferon-gamma, Dr. Denning noted.
Diagnostic criteria
The European guidelines categorize Aspergillus infections according to differences in clinical management:
• Simple aspergilloma. A single pulmonary cavity containing a fungal ball, supported by serologic or microbiologic evidence of infections with Aspergillus species in patients who are not immunocompromised and are asymptomatic or have only minor symptoms and no radiographic evidence of progression for at least 3 months.
• Chronic cavitary pulmonary aspergillosis (CCPA). The presence of one or more pulmonary cavities that may contain one or more aspergillomas or irregular intraluminal material, evidence of Aspergillus species, significant pulmonary/systemic symptoms, and overt progression on radiography over 3 or more months of observations.
• Chronic fibrosing pulmonary aspergillosis (CFPA). Severe, fibrotic destruction of at least two lung lobes as a complication of CCPA, causing a major loss of lung function. The guidelines note that destruction of a single lobe is designated as CCPA of that lobe.
• Aspergillus nodules. This unusual presentation is marked by the presence of one or more nodules that may or may not cavitate. The nodules may resemble tuberculoma, carcinoma of the lung, or coccidioidomycosis; histology is required to make an accurate diagnosis.
• Subacute invasive aspergillosis (SAIA). This can occur over the course of 1-3 months in patients who are mildly immunocompromised. Radiologic features can vary, and may include cavitation, the presence of nodules, and progressive consolidation with the appearance of abscess formation. Fungal hyphae (filaments) can be seen in biopsied lung tissues, and there may be evidence of Aspergillus galactomannan antigen in respiratory fluids or blood.
Treatment
The guidelines note that most of the evidence for managing CPA are based on cohort studies and case reports rather than randomized clinical trials, and that there have been no head-to-head trials comparing oral triazole agents.
For treatment of CPA, the European guidelines recommend:
• Itraconazole 200 mg twice daily, with therapeutic drug monitoring and dose adjustment as necessary (Grade A [strong] recommendation).
• Voriconazole 150-200 mg twice daily, with monitoring and dose adjustment. The guidelines recommend lower doses for patients older than 70 years, those with low body weight, significant liver disease, and/or those of Northeast Asian descent, who may be genetically inclined to slow drug metabolism (Grade A).
• Posaconazole liquid 400 mg twice daily, or tablets 300 mg once daily (Grade B [moderate] recommendation].
In general, the recommended duration of therapy for control of infection in patients with CPA or curative intent for patients with SAIA or chronic necrotizing pulmonary aspergillosis is 6 months or more, depending on patient status and drug tolerance.
For patients with CPA with progressive disease, those whom therapy has failed, or those who are intolerant of or have disease resistant to triazoles, intravenous therapy with micafungin, 150 mg day (Grade B); amphotericin B deoxycholate, 0.7-1.0 mg/kg per day (Grade C [marginal] recommendation); liposomal amphotericin B, 3 mg/kg per day (Grade B); or caspofungin, 50-70 mg/day (Grade C) are recommended.
The guidelines also recommend surgical excision of simple aspergilloma, preferably by a video-assisted thoracic surgery technique, if technically feasible.
“In my own experience, we resort to surgery very infrequently,” Dr. Edelman said.
He noted that it would be helpful if the guidelines had also allergic bronchopulmonary aspergillosis as a separate entity.
Ideal not always achievable
Prof. Denning points out that the optimum therapies and practices described in the guidelines can’t always be implemented. Worldwide, he said, antifungal therapy is not widely available, with the exception of fluconazole, which has no activity against Aspergillus, and is inferior to itraconazole and other extended azoles for other fungal diseases such as histoplasmosis, blastomycosis, and paracoccidioidomycosis.
The price of antifungal therapies can also be a barrier to effective treatment in many parts of the world.
“If you’re having to pay for your medicines and you’re living on $5 or $10 a day in Kenya, say, you can’t afford to buy them. So even if the drugs are physically there, it may not be really affordable for a course of therapy for these patients, and there’s some advocacy to be done around that for the whole world,” he said.
The guidelines were funded primarily by grants from ESCMID and ERS with additional support from ECMM. Authors’ travel expenses were funded jointly by ESCMID and ERS. Dr. Denning has received grant support and founder shares in F2G, and has received grants from the Fungal Research Trust, Wellcome Trust, Moulton Trust, Medical Research Council, Chronic Granulomatous Disease Research Trust, National Institute of Allergy and Infectious Diseases, National Institute of Health Research and the European Union, and AstraZeneca. Dr. Edelman reported no relevant disclosures.
FROM JOURNAL OF DRUGS IN DERMATOLOGY
Chemo quadruples risk for myeloid cancers
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.

“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
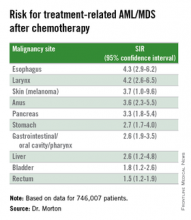
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.
“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
ORLANDO – Patients who undergo cytotoxic chemotherapy, even in the modern era, are at increased risk for developing myeloid neoplasms, based on data from a cohort of nearly 750,000 adults who were initially treated with chemotherapy and survived at least 1 year after diagnosis.
In the cohort, the standardized incidence ratio (SIR) for treatment-related acute myeloid leukemia (tAML) or myelodysplastic syndrome (MDS) was four times greater than would be expected in the general population, reported Lindsay M. Morton, Ph.D., of the division of cancer epidemiology and genetics at the National Cancer Institute in Bethesda, Md.
“We now demonstrate that there are elevated risks for treatment-related leukemia after treatment for a broad spectrum of first primary malignancies that are generally consistent with what we know about changing treatment practices,” she said at the American Society of Hematology annual meeting.
“The number of cancer survivors in the United States has increased dramatically, to reach nearly 14 million individuals today, and in just the next few years the number is expected to reach more than 18 million people, which means that the long-term health of this population is of great clinical importance as well as public health importance,” Dr. Morton emphasized.
The link between cytotoxic chemotherapy and leukemia risk has been known since the 1960s, with certain classes of agents carrying higher risks than others, including platinum-containing compounds, alkylating agents (for example, cyclophosphamide, melphalan, chlorambucil), topoisomerase II inhibitors (doxorubicin, daunorubicin, epirubicin, etc.), and antimetabolites (5-fluorauracil, capecitabine, gemcitabine, et al.).
Treatment-related leukemias are associated with higher doses of these agents, and the trend in contemporary medicine is to use more of these agents upfront for the treatment of primary malignancies. Yet estimates of the risk of tAML, MDS, or other malignancies have been hard to come by because of the relative rarity of cases and the sporadic reports in the literature, Dr. Morton said.
The investigators previously reported that risk for tAML and other myeloid neoplasms changed over time, and showed that since the 1990s there was an uptick in risk for patients treated with chemotherapy for cancers of bone, joints, and endometrium, and since 2000 for patients treated with chemotherapy for cancers of the esophagus, cervix and prostate.
For example, risks for tAML were higher in the 1970s for patients with ovarian cancer treated with melphalan, a highly leukemogenic agent, but dropped somewhat with the switch to platinum-based agents. Similarly, women with breast cancer had a comparatively high risk with the use of melphalan, a decline in risk with the introduction of cyclophosphamide, and then an increase with the addition of anthracyclines and dose-dense regimens.
Risk update
To get a better idea of the magnitude of risk in the modern era, Dr. Morton and colleagues sifted through Surveillance, Epidemiology, and End Results (SEER) data to identify a cohort of 746,007 adults who were initially treated with chemotherapy and survived for at least 1 year following a diagnosis with a first primary malignancy from 2000 through 2012. They calculated SIRs based on variables that included age, race, sex, malignancy type and treatment period.
They looked at four categories of myeloid neoplasms as defined by World Health Organization criteria: AML/MDS, chronic myeloid leukemia, myeloproliferative neoplasms (MPN) negative for BCR-ABL (Philadelphia-negative), and chronic myelomonocytic leukemia (CMML).
They found that 2,071 patients developed treatment-related AML/MDS, translating into a fourfold incidence compared with the general population (SIR 4.1, 95% confidence interval [CI] 3.9-4.2), 106 were diagnosed with CMML
They also identified novel risk for tAML/MDS after chemotherapy by malignancy (see table).
The investigators found that breast cancer, non-Hodgkin lymphoma, and lung cancer were most commonly associated with tAML/MDS (SIRs 4.1, 7.3, and 4.1, respectively, all significant).
In addition, although the overall numbers of cases were small, the investigators noted “strikingly elevated” risks for cancers of bone (SIR 35.1, CI. 16.9-64.6). testes (15.6, CI, 9.2-24.6), and soft tissue (12.6, CI=7.7-19.4),
Risk for tAML/MD was more modestly elevated for cancers of the brain, ovaries, endometrium, cervix, and prostate, and for Hodgkin lymphoma, chronic lymphocytic leukemia, and multiple myeloma.
Adding radiotherapy to chemotherapy for cancers of the breast, lung, and stomach cancers showed a trend toward heightened tAML/MDS risk, but this was not significant.
An elevated risk for CMML was also seen after chemotherapy for lung cancer (SIR 2.5, CI, 1.3-4.4), breast cancer (1.8, CI, 1.3-2.5), and non-Hodgkin lymphoma (2.1, CI, 1.2-3.4). There was elevated risk for CMML following chemotherapy for breast cancer (3.0, CI. 1.7-5.0) and non-Hodgkin lymphoma (4.2, CI, 2.4-6.9).
There were no increased risks for other myeloproliferative neoplasms after chemotherapy for any first primary cancer, however.
“This reminds us that with new uses of standard agents and introduction of new agents, it’s critical to carefully weigh the risks and benefits of systemic therapy,” Dr. Morton said.
The investigators plan to quantify risks associated with specific drugs and doses, she added.
The study was supported by the National Cancer Institute. Dr. Morton reported no relevant conflicts of interest to disclose.
AT ASH 2015
<p><b>Key clinical point: </b>This study quantifies the risks for treatment-related myeloid cancers after chemotherapy.
</p><p><b>Major finding: </b>Chemotherapy is associated with a fourfold risk for treatment-related AML/MDS, compared with the general population.
</p><p><b>Data source: </b>Retrospective review of data on 746,007 adults treated with chemotherapy for a first primary malignancy.
</p><p><b>Disclosures: </b>The National Cancer Institute supported the study. Dr. Morton reported having no conflicts of interest to disclose.</p>

Anti-BCL2, CD20 combo safe, effective in untreated CLL
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.

“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.
“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
ORLANDO – An experimental combination of obinutuzumab and venetoclax appears safe as frontline therapy for patients with active, untreated chronic lymphocytic leukemia and comorbidities.
In the safety run-in portion of a phase 3, open-label trial comparing the combination of obinutuzumab (Gazyva) and the investigational Bcl-2 inhibitor venetoclax with obinutuzumab and its usual partner chlorambucil in 12 patients, only two of seven patients classified as being at high risk for the tumor lysis syndrome (TLS) developed laboratory-defined TLS, and no patients had clinical TLS.
The combination did not meet any of the pre-specified stopping criteria, and early data hinted at efficacy for the combination, said Dr. Kirsten Fischer, from the Center for Integrated Oncology at University Hospital Cologne in Germany.
“As with previous reports, our data confirm rapid and profound reduction in lymphocyte counts after the first dose obinutuzumab in all 12 [evaluable] patients,” she said at the American Society of Hematology annual meeting.
In the CLL 11 trial, investigators in the German CLL group previously showed that the combination of the anti-CD20 antibody obinutuzumab and chlorambucil resulted in improved overall survival compared to chlorambucil alone in patients with previously untreated CLL and coexisting medical conditions. The combination is approved in the United States for adults with treatment-naïve CLL.
Venetoclax has been shown to have good efficacy against relapsed/refractory CLL both as monotherapy and in combination with rituximab, prompting the investigators to explore it in combination with the rituximab follow-on drug obinutuzumab.
In the safety run-in phase of the CLL 14 trial, the investigators enrolled 13 adults (median age 75, range 59-88 years) with newly diagnosed, active confirmed CLL and coexisting medical conditions as determined either by a score greater 6 on the cumulative illness rating scale (CIRS) or by estimated creatinine clearance less than 70 mL/min.
The patients were treated with 6 cycles of obinutuzumab and venetoclax followed by 6 additional cycles of venetoclax. Obinutuzumab was administered intravenously 100 mg on day 1 and 900 mg on day 2, with the option to deliver 1,000 mg on day 1 instead, then 1,000 mg on days 8 and 15 of cycle 1, and 1,000 mg on day 1 for cycles 2-6.
The dose of venetoclax was titrated upward gradually, with doses of 20 mg, 50 mg, 100 mg, 200 mg, and up to 400 mg administered starting on day 22 of cycle 1.
Planned stopping criteria were one treatment-related death or a grade 4 adverse event related to clinical TLS despite prophylaxis as specified by the protocol.
At the time of data cutoff (October 2015), 12 patients had been on treatment for at least 4 weeks and had reached the maximum venetoclax dose. Two patients had reached 11 cycles, three had reached 10 cycles, and seven had reached 8 cycles.
Grade 1 or 2 adverse events in all 13 enrolled patients included infusion-related reactions in 8; infections in 6; diarrhea, hyperkalemia, and constipation in 5; nausea, dizziness and cough in 4; and fatigue, headache and pruritus in 3.
Grade 3 or 4 adverse events were neutropenia in five patients; infusion related reactions; syncope, thrombocytopenia and laboratory-defined TLS in two patients; and bradycardia, hyperglycemia, influenza, leucopenia, pyrexia, respiratory tract infection, and elevated transaminases in one patient each.
As noted, all 12 evaluable patients had rapid drops in absolute lymphocyte counts, and all but one had complete resolution of lymphadenopathy after three cycles, with the improvement maintained after six cycles. The remaining patient had a decrease to near normal after both three and six cycles.
Of the 12 patients, 11 had a partial response after three cycles, and the remaining patient had stable disease, for an overall response rate of 92%. The overall response rate after six cycles was 100%, with all patients having a partial response.
The data were sufficiently good to justify continuing with the randomized phase, which began in August 2015, Dr. Fischer noted.
The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.
AT ASH 2015
<p><b>Key clinical point:</b> Patients with CLL and comorbidities were able to tolerate an experimental regimen of obinutuzumab and venetoclax.
</p><p><b>Major finding:</b> Two of 12 evaluable patients had evidence of laboratory-defined but not clinical tumor lysis syndrome.
</p><p><b>Data source:</b> Safety run-in phase of a randomized phase 3 trial with 13 patients with chronic lymphocytic leukemia and comorbidities.
</p><p><b>Disclosures:</b> The study is sponsored by Hoffman-La Roche and AbbVie. Dr. Fischer disclosed receiving travel grants from Hoffman-La Roche.</p>

ASH: Idelalisib plus standard therapy boosts survival in relapsed CLL
ORLANDO – Adding the PI3K inhibitor idelalisib to a standard regimen of bendamustine and rituximab significantly reduced the risk of both disease progression and death for patients with relapsed and/or refractory chronic lymphocytic leukemia, results of a phase III randomized trial showed.
At a median follow-up of 12 months, the primary endpoint of median progression-free survival was 23.1 months for patients treated with idelalisib (Zydelig), bendamustine, and rituximab (idel+BR), compared with 11.1 months for bendamustine and rituximab (BR) plus a placebo, reported Dr. Andrew Zelenetz of Memorial Sloan Kettering Cancer Center, New York.

“Median overall survival was not reached in either arm. However, there was a significant improvement in overall survival, with a 45% reduction in the risk of death [with idel+BR],” he said in a late-breaking abstract session at the annual meeting of the American Society of Hematology.
The trial was stopped early after a data review at the first planned interim analysis showed significant superiority for the three-drug combination.
The results were consistent across subgroups, including patients with high-risk features such as deletion 17p and mutated TP53 (del[17p]/TP53), unmutated immunoglobulin heavy chain variable region (IgHV), and treatment-refractory disease.
The rationale behind adding idelalisib, an inhibitor of the phosphatidylinositol-3 kinase (PI3K), is that signaling via the PI3K pathway is hyperactive and can be targeted, Dr. Zelenetz explained.
Study 115 was a phase III trial with accrual from June 2012 through August 2015. Investigators enrolled 416 patients with relapsed /refractory CLL and randomly assigned them to receive BR for six 28-day cycles of bendamustine (70 mg/m2 on days 1 and 2 of each cycle) and rituximab (375 mg/m2 for cycle 1, and 500 mg/m2 for cycles 2 through 6), plus either idelalisib 150 mg b.i.d. or placebo, each administered continuously until disease progression, intolerable toxicity, withdrawal of consent, or death.
The patients were stratified by mutational and disease status (refractory defined as CLL progression less than 6 months from completion of prior therapy, or relapsed CLL progression 6 months or more from completion of prior therapy.
The trial was halted early after the first planned interim analysis, which was conducted after 75% of the total number of 260 planned events of CLL progression or death from any cause had occurred. The data cutoff was June 15, 2015.
The intention-to-treat analysis included 207 patients assigned to idelalisib and 209 assigned to placebo. Three-fourths (76%) of the patients were male.
In all, 46% of patients had Rai stage III/IV disease. The median time since the completion of the last therapy was 16 months.
The proportions of patients with high-risk features included del(17p)/p53 mutation in 32.9%, unmutated IgHV in 83.2%, and treatment-refractory disease in 29.8%.
As noted, the median progression-free survival with idelalisib at a median follow-up of 12 months was 23.1 months vs. 11.1 months for placebo. That translated into a hazard ratio of 0.33 (P less than .0001).
Among patients with neither del(17p) nor TP53 mutations, the HR for progression was 0.22. Among patients with either del(17p) or a TP53 mutation, the HR was 0.50 (95% confidence intervals show statistical significance for both).
Overall response rates were 68% among patients who received idelalisib, and 45% for those who received placebo. There were five complete responses (2%) in the idelalisib group and none in the placebo group.
The idelalisib group also had a higher proportion of patients with a greater than 50% reduction in involved lymph nodes (96% vs. 61%), and had better organomegaly responses (spleen and liver) and hematologic responses (hemoglobin, neutrophils, and platelets).
Grade 3 or greater adverse events occurred in 93% of patients on idelalisib, compared with 76% of those on placebo. The proportion of patients with any serious adverse event was 66% vs. 44%, respectively.
Adverse events leading to drug dose reduction were seen in 11% of idelalisib-treated patients, compared with 6% of placebo controls, and therapy was discontinued in 26% vs. 13%, respectively.
Ten patients in the idelalisib arm and seven in the placebo arm died during the study.
Adverse events that occurred more commonly with idelalisib included neutropenia, pyrexia, diarrhea, febrile neutropenia, pneumonia, rash, and elevated liver enzymes.
Session moderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston asked Dr. Zelenetz how idelalisib plus BR stacked up to ibrutinib (Imbruvica) plus BR in this population.
Dr. Zelenetz noted that patients were excluded from the HELIOS trial of ibrutinib plus BR if they had del(17p). Comparing the subset of patients in Study 115 without del(17p) with patients in the ibrutinib study, “the results are virtually superimposable,” Dr. Zelenetz said, and “the two treatments are really remarkably similar.”
The overall survival benefit was larger in the HELIOS trial, Dr. Zelenetz noted, but that was largely because the trial allowed patients to cross over from placebo to the active drug.
Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.
ORLANDO – Adding the PI3K inhibitor idelalisib to a standard regimen of bendamustine and rituximab significantly reduced the risk of both disease progression and death for patients with relapsed and/or refractory chronic lymphocytic leukemia, results of a phase III randomized trial showed.
At a median follow-up of 12 months, the primary endpoint of median progression-free survival was 23.1 months for patients treated with idelalisib (Zydelig), bendamustine, and rituximab (idel+BR), compared with 11.1 months for bendamustine and rituximab (BR) plus a placebo, reported Dr. Andrew Zelenetz of Memorial Sloan Kettering Cancer Center, New York.
“Median overall survival was not reached in either arm. However, there was a significant improvement in overall survival, with a 45% reduction in the risk of death [with idel+BR],” he said in a late-breaking abstract session at the annual meeting of the American Society of Hematology.
The trial was stopped early after a data review at the first planned interim analysis showed significant superiority for the three-drug combination.
The results were consistent across subgroups, including patients with high-risk features such as deletion 17p and mutated TP53 (del[17p]/TP53), unmutated immunoglobulin heavy chain variable region (IgHV), and treatment-refractory disease.
The rationale behind adding idelalisib, an inhibitor of the phosphatidylinositol-3 kinase (PI3K), is that signaling via the PI3K pathway is hyperactive and can be targeted, Dr. Zelenetz explained.
Study 115 was a phase III trial with accrual from June 2012 through August 2015. Investigators enrolled 416 patients with relapsed /refractory CLL and randomly assigned them to receive BR for six 28-day cycles of bendamustine (70 mg/m2 on days 1 and 2 of each cycle) and rituximab (375 mg/m2 for cycle 1, and 500 mg/m2 for cycles 2 through 6), plus either idelalisib 150 mg b.i.d. or placebo, each administered continuously until disease progression, intolerable toxicity, withdrawal of consent, or death.
The patients were stratified by mutational and disease status (refractory defined as CLL progression less than 6 months from completion of prior therapy, or relapsed CLL progression 6 months or more from completion of prior therapy.
The trial was halted early after the first planned interim analysis, which was conducted after 75% of the total number of 260 planned events of CLL progression or death from any cause had occurred. The data cutoff was June 15, 2015.
The intention-to-treat analysis included 207 patients assigned to idelalisib and 209 assigned to placebo. Three-fourths (76%) of the patients were male.
In all, 46% of patients had Rai stage III/IV disease. The median time since the completion of the last therapy was 16 months.
The proportions of patients with high-risk features included del(17p)/p53 mutation in 32.9%, unmutated IgHV in 83.2%, and treatment-refractory disease in 29.8%.
As noted, the median progression-free survival with idelalisib at a median follow-up of 12 months was 23.1 months vs. 11.1 months for placebo. That translated into a hazard ratio of 0.33 (P less than .0001).
Among patients with neither del(17p) nor TP53 mutations, the HR for progression was 0.22. Among patients with either del(17p) or a TP53 mutation, the HR was 0.50 (95% confidence intervals show statistical significance for both).
Overall response rates were 68% among patients who received idelalisib, and 45% for those who received placebo. There were five complete responses (2%) in the idelalisib group and none in the placebo group.
The idelalisib group also had a higher proportion of patients with a greater than 50% reduction in involved lymph nodes (96% vs. 61%), and had better organomegaly responses (spleen and liver) and hematologic responses (hemoglobin, neutrophils, and platelets).
Grade 3 or greater adverse events occurred in 93% of patients on idelalisib, compared with 76% of those on placebo. The proportion of patients with any serious adverse event was 66% vs. 44%, respectively.
Adverse events leading to drug dose reduction were seen in 11% of idelalisib-treated patients, compared with 6% of placebo controls, and therapy was discontinued in 26% vs. 13%, respectively.
Ten patients in the idelalisib arm and seven in the placebo arm died during the study.
Adverse events that occurred more commonly with idelalisib included neutropenia, pyrexia, diarrhea, febrile neutropenia, pneumonia, rash, and elevated liver enzymes.
Session moderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston asked Dr. Zelenetz how idelalisib plus BR stacked up to ibrutinib (Imbruvica) plus BR in this population.
Dr. Zelenetz noted that patients were excluded from the HELIOS trial of ibrutinib plus BR if they had del(17p). Comparing the subset of patients in Study 115 without del(17p) with patients in the ibrutinib study, “the results are virtually superimposable,” Dr. Zelenetz said, and “the two treatments are really remarkably similar.”
The overall survival benefit was larger in the HELIOS trial, Dr. Zelenetz noted, but that was largely because the trial allowed patients to cross over from placebo to the active drug.
Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.
ORLANDO – Adding the PI3K inhibitor idelalisib to a standard regimen of bendamustine and rituximab significantly reduced the risk of both disease progression and death for patients with relapsed and/or refractory chronic lymphocytic leukemia, results of a phase III randomized trial showed.
At a median follow-up of 12 months, the primary endpoint of median progression-free survival was 23.1 months for patients treated with idelalisib (Zydelig), bendamustine, and rituximab (idel+BR), compared with 11.1 months for bendamustine and rituximab (BR) plus a placebo, reported Dr. Andrew Zelenetz of Memorial Sloan Kettering Cancer Center, New York.
“Median overall survival was not reached in either arm. However, there was a significant improvement in overall survival, with a 45% reduction in the risk of death [with idel+BR],” he said in a late-breaking abstract session at the annual meeting of the American Society of Hematology.
The trial was stopped early after a data review at the first planned interim analysis showed significant superiority for the three-drug combination.
The results were consistent across subgroups, including patients with high-risk features such as deletion 17p and mutated TP53 (del[17p]/TP53), unmutated immunoglobulin heavy chain variable region (IgHV), and treatment-refractory disease.
The rationale behind adding idelalisib, an inhibitor of the phosphatidylinositol-3 kinase (PI3K), is that signaling via the PI3K pathway is hyperactive and can be targeted, Dr. Zelenetz explained.
Study 115 was a phase III trial with accrual from June 2012 through August 2015. Investigators enrolled 416 patients with relapsed /refractory CLL and randomly assigned them to receive BR for six 28-day cycles of bendamustine (70 mg/m2 on days 1 and 2 of each cycle) and rituximab (375 mg/m2 for cycle 1, and 500 mg/m2 for cycles 2 through 6), plus either idelalisib 150 mg b.i.d. or placebo, each administered continuously until disease progression, intolerable toxicity, withdrawal of consent, or death.
The patients were stratified by mutational and disease status (refractory defined as CLL progression less than 6 months from completion of prior therapy, or relapsed CLL progression 6 months or more from completion of prior therapy.
The trial was halted early after the first planned interim analysis, which was conducted after 75% of the total number of 260 planned events of CLL progression or death from any cause had occurred. The data cutoff was June 15, 2015.
The intention-to-treat analysis included 207 patients assigned to idelalisib and 209 assigned to placebo. Three-fourths (76%) of the patients were male.
In all, 46% of patients had Rai stage III/IV disease. The median time since the completion of the last therapy was 16 months.
The proportions of patients with high-risk features included del(17p)/p53 mutation in 32.9%, unmutated IgHV in 83.2%, and treatment-refractory disease in 29.8%.
As noted, the median progression-free survival with idelalisib at a median follow-up of 12 months was 23.1 months vs. 11.1 months for placebo. That translated into a hazard ratio of 0.33 (P less than .0001).
Among patients with neither del(17p) nor TP53 mutations, the HR for progression was 0.22. Among patients with either del(17p) or a TP53 mutation, the HR was 0.50 (95% confidence intervals show statistical significance for both).
Overall response rates were 68% among patients who received idelalisib, and 45% for those who received placebo. There were five complete responses (2%) in the idelalisib group and none in the placebo group.
The idelalisib group also had a higher proportion of patients with a greater than 50% reduction in involved lymph nodes (96% vs. 61%), and had better organomegaly responses (spleen and liver) and hematologic responses (hemoglobin, neutrophils, and platelets).
Grade 3 or greater adverse events occurred in 93% of patients on idelalisib, compared with 76% of those on placebo. The proportion of patients with any serious adverse event was 66% vs. 44%, respectively.
Adverse events leading to drug dose reduction were seen in 11% of idelalisib-treated patients, compared with 6% of placebo controls, and therapy was discontinued in 26% vs. 13%, respectively.
Ten patients in the idelalisib arm and seven in the placebo arm died during the study.
Adverse events that occurred more commonly with idelalisib included neutropenia, pyrexia, diarrhea, febrile neutropenia, pneumonia, rash, and elevated liver enzymes.
Session moderator Dr. David P. Steensma of the Dana-Farber Cancer Institute in Boston asked Dr. Zelenetz how idelalisib plus BR stacked up to ibrutinib (Imbruvica) plus BR in this population.
Dr. Zelenetz noted that patients were excluded from the HELIOS trial of ibrutinib plus BR if they had del(17p). Comparing the subset of patients in Study 115 without del(17p) with patients in the ibrutinib study, “the results are virtually superimposable,” Dr. Zelenetz said, and “the two treatments are really remarkably similar.”
The overall survival benefit was larger in the HELIOS trial, Dr. Zelenetz noted, but that was largely because the trial allowed patients to cross over from placebo to the active drug.
Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.
AT ASH 2015
<p><b>Key clinical point:</b> Adding the PI3K inhibitor idelalisib to bendamustine and rituximab significantly improved survival of patients with relapsed/refractory CLL.
</p><p><b>Major finding: </b>Median progression-free survival was 23.1 months for patients treated with idelalisib, bendamustine, and rituximab, compared with 11.1 months for bendamustine and rituximab plus placebo.
</p><p><b>Data source:</b> Randomized, controlled trial in 416 patients with relapsed/refractory CLL. The trial was halted early for superior efficacy with idelalisib.
</p><p><b>Disclosures: </b>Gilead Sciences funded Study 115. Dr. Zelenetz disclosed receiving research funding from the company and discussing off-label use of idelalisib for relapsed/refractory CLL.</p>

Upfront idelalisib carries high risk for acute liver toxicity
ORLANDO – Idelalisib given as first-line therapy for patients with chronic lymphocytic leukemia carries a high risk of early fulminant hepatotoxicity requiring drug interruption and steroids, investigators reported.
Among 24 patients who received idelalisib (Zydelig) monotherapy in a phase II trial of a combination of idelalisib followed by idelalisib concurrent with ofatumumab (Arzerra) as first-line therapy for chronic lymphocytic leukemia (CLL), 12 patients developed acute hepatotoxicity, marked by rapidly soaring levels of transaminase within about 28 days of starting therapy. An additional four patients developed hepatotoxicity at around 130 days, noted Dr. Benjamin L. Lampson, a clinical fellow in medicine at the Dana-Farber Cancer Institute in Boston.

“Multiple lines of evidence suggest that this early hepatotoxicity is immune mediated. The proportion of regulatory T cells in the peripheral blood decrease on idelalisib therapy, providing a possible explanation for the development of early hepatotoxicity,” he said at the annual meeting of the American Society of Hematology.
The toxicities occur more frequently among younger and less heavily pretreated patients, and are likely due to on-target, immune-mediated effects, he noted.
Dr. Lampson presented data on the first 24 patients in an ongoing phase II trial. Patients with previously untreated CLL receive idelalisib 150 mg twice daily for 56 days, in an attempt to mobilize neoplastic B cells from the peripheral lymphoid tissues and into the bloodstream.
Following the monotherapy phase, patients are given ofatumumab in an attempt to clear the disease from peripheral blood.
“This dosing strategy is slightly different than what has been previously been used in trials combining these particular drugs. Specifically, previously reported trials have started these agents simultaneously without a lead-in period of monotherapy,” Dr. Lampson explained.
When the lead-in phase is completed, patients receive idelalisib plus ofatumumab infusions once weekly for 8 weeks, followed by once-monthly infusions for 4 months. Patients then continue on idelalisib indefinitely. The primary endpoint is the overall response rate assessed 2 months after the completion of the combination therapy.
For the first 24 patients treated as of Nov. 9, 2015, the median time on therapy was 7.7 months and median follow-up was 14.7 months.
The median patient age was 67.4 years (range 57.6-84.9). CLL genetics showed that 13 patients had unmutated immunoglobulin heavy chain variable region (IgHV) disease, 4 had the 17p deletion and TP53 mutation, 1 had deletion 11q, and 13 had deletion 13q; some patients had more than one mutation.
“What we began to notice after enrolling just a few subjects on the trial was that severe hepatotoxicity was occurring shortly after initiating idelalisib,” Dr. Lampson said.
He presented one case, a 58-year-old man who was in the idelalisib monotherapy phase of the study. He developed grade 3 hepatotoxicity 28 days after starting the drug, despite having a normal liver function test just 1 day earlier. The drug was stopped, but his liver function tests continued to rise, suggesting a self-perpetuating or self-sustaining process.
On day 32, the patient was admitted to the hospital, and on day 33 he was started on steroids, based on the hypothesis that the hepatotoxicity might have been immune mediated. Two days after initiation of steroids, his liver function tests continued to rise, whereupon he was started on mycophenolate mofetil.
“With these two forms of immunosuppression, the [liver function tests] did eventually normalize, although the steroids and mycophenolate had to be tapered over a period of many weeks. And this patient was not the only patient to experience toxicity; in fact, hepatotoxicity was frequent and often severe,” he said.
At the time of maximum incidence, week 4, the percentage of patients with any hepatotoxicity was 46%, with 13% at grade 4, and 21% at grade 3.
“The median time to initial development of hepatotoxicity is 28 days. This suggests that the mechanism of hepatotoxicity is not immediate, but takes time to develop, consistent with an adaptive immune response. Furthermore, hepatotoxicity is typically occurring before the first dose of ofatumumab is occurring at week 8, suggesting idelalisib alone is the cause of the hepatotoxicity,” Dr. Lampson said.
A comparison of data from the ongoing study and from three previous studies – two with idelalisib in relapsed refractory disease, and one as first-line therapy in patients 65 and older – showed that grade 3 or greater hepatotoxicity was lowest in a phase I trial of idelalisib in which patients had received a median of five prior lines of therapy, occurring in only 1.9% of patients. In contrast, in the current study, 52% of patients experienced grade 3 or 4 transaminitis at some point in the trial.
Evidence for the hepatotoxicity being an on-target immune-mediated effect comes from lymphocytic infiltrate on liver biopsy and lymphocytic colitis in idelalisib-treated patients. Additional evidence comes from the fact that the toxicity is both treatable and preventable with steroids, he said.
He cautioned that hepatotoxicity can recur rapidly when the drug is reintroduced.
“In general, our experience has been if idelalisib is resumed while the subject remains on steroids, the drug is more likely to be tolerated and the subject eventually can be tapered off steroids,” he said.
Asked by an audience member whether patients who are receiving idelalisib in the first-line setting should also receive steroids, Dr. Lampson said that they closely monitor patient liver enzymes around 28 days, and if grade 1 transaminitis is detected, patients are automatically started on low-dose steroids.
The study is sponsored by the Dana-Farber Cancer Institute in collaboration with Gilead Sciences and GlaxoSmithKline. Dr. Lampson and colleagues declared no relevant conflicts of interest.
ORLANDO – Idelalisib given as first-line therapy for patients with chronic lymphocytic leukemia carries a high risk of early fulminant hepatotoxicity requiring drug interruption and steroids, investigators reported.
Among 24 patients who received idelalisib (Zydelig) monotherapy in a phase II trial of a combination of idelalisib followed by idelalisib concurrent with ofatumumab (Arzerra) as first-line therapy for chronic lymphocytic leukemia (CLL), 12 patients developed acute hepatotoxicity, marked by rapidly soaring levels of transaminase within about 28 days of starting therapy. An additional four patients developed hepatotoxicity at around 130 days, noted Dr. Benjamin L. Lampson, a clinical fellow in medicine at the Dana-Farber Cancer Institute in Boston.
“Multiple lines of evidence suggest that this early hepatotoxicity is immune mediated. The proportion of regulatory T cells in the peripheral blood decrease on idelalisib therapy, providing a possible explanation for the development of early hepatotoxicity,” he said at the annual meeting of the American Society of Hematology.
The toxicities occur more frequently among younger and less heavily pretreated patients, and are likely due to on-target, immune-mediated effects, he noted.
Dr. Lampson presented data on the first 24 patients in an ongoing phase II trial. Patients with previously untreated CLL receive idelalisib 150 mg twice daily for 56 days, in an attempt to mobilize neoplastic B cells from the peripheral lymphoid tissues and into the bloodstream.
Following the monotherapy phase, patients are given ofatumumab in an attempt to clear the disease from peripheral blood.
“This dosing strategy is slightly different than what has been previously been used in trials combining these particular drugs. Specifically, previously reported trials have started these agents simultaneously without a lead-in period of monotherapy,” Dr. Lampson explained.
When the lead-in phase is completed, patients receive idelalisib plus ofatumumab infusions once weekly for 8 weeks, followed by once-monthly infusions for 4 months. Patients then continue on idelalisib indefinitely. The primary endpoint is the overall response rate assessed 2 months after the completion of the combination therapy.
For the first 24 patients treated as of Nov. 9, 2015, the median time on therapy was 7.7 months and median follow-up was 14.7 months.
The median patient age was 67.4 years (range 57.6-84.9). CLL genetics showed that 13 patients had unmutated immunoglobulin heavy chain variable region (IgHV) disease, 4 had the 17p deletion and TP53 mutation, 1 had deletion 11q, and 13 had deletion 13q; some patients had more than one mutation.
“What we began to notice after enrolling just a few subjects on the trial was that severe hepatotoxicity was occurring shortly after initiating idelalisib,” Dr. Lampson said.
He presented one case, a 58-year-old man who was in the idelalisib monotherapy phase of the study. He developed grade 3 hepatotoxicity 28 days after starting the drug, despite having a normal liver function test just 1 day earlier. The drug was stopped, but his liver function tests continued to rise, suggesting a self-perpetuating or self-sustaining process.
On day 32, the patient was admitted to the hospital, and on day 33 he was started on steroids, based on the hypothesis that the hepatotoxicity might have been immune mediated. Two days after initiation of steroids, his liver function tests continued to rise, whereupon he was started on mycophenolate mofetil.
“With these two forms of immunosuppression, the [liver function tests] did eventually normalize, although the steroids and mycophenolate had to be tapered over a period of many weeks. And this patient was not the only patient to experience toxicity; in fact, hepatotoxicity was frequent and often severe,” he said.
At the time of maximum incidence, week 4, the percentage of patients with any hepatotoxicity was 46%, with 13% at grade 4, and 21% at grade 3.
“The median time to initial development of hepatotoxicity is 28 days. This suggests that the mechanism of hepatotoxicity is not immediate, but takes time to develop, consistent with an adaptive immune response. Furthermore, hepatotoxicity is typically occurring before the first dose of ofatumumab is occurring at week 8, suggesting idelalisib alone is the cause of the hepatotoxicity,” Dr. Lampson said.
A comparison of data from the ongoing study and from three previous studies – two with idelalisib in relapsed refractory disease, and one as first-line therapy in patients 65 and older – showed that grade 3 or greater hepatotoxicity was lowest in a phase I trial of idelalisib in which patients had received a median of five prior lines of therapy, occurring in only 1.9% of patients. In contrast, in the current study, 52% of patients experienced grade 3 or 4 transaminitis at some point in the trial.
Evidence for the hepatotoxicity being an on-target immune-mediated effect comes from lymphocytic infiltrate on liver biopsy and lymphocytic colitis in idelalisib-treated patients. Additional evidence comes from the fact that the toxicity is both treatable and preventable with steroids, he said.
He cautioned that hepatotoxicity can recur rapidly when the drug is reintroduced.
“In general, our experience has been if idelalisib is resumed while the subject remains on steroids, the drug is more likely to be tolerated and the subject eventually can be tapered off steroids,” he said.
Asked by an audience member whether patients who are receiving idelalisib in the first-line setting should also receive steroids, Dr. Lampson said that they closely monitor patient liver enzymes around 28 days, and if grade 1 transaminitis is detected, patients are automatically started on low-dose steroids.
The study is sponsored by the Dana-Farber Cancer Institute in collaboration with Gilead Sciences and GlaxoSmithKline. Dr. Lampson and colleagues declared no relevant conflicts of interest.
ORLANDO – Idelalisib given as first-line therapy for patients with chronic lymphocytic leukemia carries a high risk of early fulminant hepatotoxicity requiring drug interruption and steroids, investigators reported.
Among 24 patients who received idelalisib (Zydelig) monotherapy in a phase II trial of a combination of idelalisib followed by idelalisib concurrent with ofatumumab (Arzerra) as first-line therapy for chronic lymphocytic leukemia (CLL), 12 patients developed acute hepatotoxicity, marked by rapidly soaring levels of transaminase within about 28 days of starting therapy. An additional four patients developed hepatotoxicity at around 130 days, noted Dr. Benjamin L. Lampson, a clinical fellow in medicine at the Dana-Farber Cancer Institute in Boston.
“Multiple lines of evidence suggest that this early hepatotoxicity is immune mediated. The proportion of regulatory T cells in the peripheral blood decrease on idelalisib therapy, providing a possible explanation for the development of early hepatotoxicity,” he said at the annual meeting of the American Society of Hematology.
The toxicities occur more frequently among younger and less heavily pretreated patients, and are likely due to on-target, immune-mediated effects, he noted.
Dr. Lampson presented data on the first 24 patients in an ongoing phase II trial. Patients with previously untreated CLL receive idelalisib 150 mg twice daily for 56 days, in an attempt to mobilize neoplastic B cells from the peripheral lymphoid tissues and into the bloodstream.
Following the monotherapy phase, patients are given ofatumumab in an attempt to clear the disease from peripheral blood.
“This dosing strategy is slightly different than what has been previously been used in trials combining these particular drugs. Specifically, previously reported trials have started these agents simultaneously without a lead-in period of monotherapy,” Dr. Lampson explained.
When the lead-in phase is completed, patients receive idelalisib plus ofatumumab infusions once weekly for 8 weeks, followed by once-monthly infusions for 4 months. Patients then continue on idelalisib indefinitely. The primary endpoint is the overall response rate assessed 2 months after the completion of the combination therapy.
For the first 24 patients treated as of Nov. 9, 2015, the median time on therapy was 7.7 months and median follow-up was 14.7 months.
The median patient age was 67.4 years (range 57.6-84.9). CLL genetics showed that 13 patients had unmutated immunoglobulin heavy chain variable region (IgHV) disease, 4 had the 17p deletion and TP53 mutation, 1 had deletion 11q, and 13 had deletion 13q; some patients had more than one mutation.
“What we began to notice after enrolling just a few subjects on the trial was that severe hepatotoxicity was occurring shortly after initiating idelalisib,” Dr. Lampson said.
He presented one case, a 58-year-old man who was in the idelalisib monotherapy phase of the study. He developed grade 3 hepatotoxicity 28 days after starting the drug, despite having a normal liver function test just 1 day earlier. The drug was stopped, but his liver function tests continued to rise, suggesting a self-perpetuating or self-sustaining process.
On day 32, the patient was admitted to the hospital, and on day 33 he was started on steroids, based on the hypothesis that the hepatotoxicity might have been immune mediated. Two days after initiation of steroids, his liver function tests continued to rise, whereupon he was started on mycophenolate mofetil.
“With these two forms of immunosuppression, the [liver function tests] did eventually normalize, although the steroids and mycophenolate had to be tapered over a period of many weeks. And this patient was not the only patient to experience toxicity; in fact, hepatotoxicity was frequent and often severe,” he said.
At the time of maximum incidence, week 4, the percentage of patients with any hepatotoxicity was 46%, with 13% at grade 4, and 21% at grade 3.
“The median time to initial development of hepatotoxicity is 28 days. This suggests that the mechanism of hepatotoxicity is not immediate, but takes time to develop, consistent with an adaptive immune response. Furthermore, hepatotoxicity is typically occurring before the first dose of ofatumumab is occurring at week 8, suggesting idelalisib alone is the cause of the hepatotoxicity,” Dr. Lampson said.
A comparison of data from the ongoing study and from three previous studies – two with idelalisib in relapsed refractory disease, and one as first-line therapy in patients 65 and older – showed that grade 3 or greater hepatotoxicity was lowest in a phase I trial of idelalisib in which patients had received a median of five prior lines of therapy, occurring in only 1.9% of patients. In contrast, in the current study, 52% of patients experienced grade 3 or 4 transaminitis at some point in the trial.
Evidence for the hepatotoxicity being an on-target immune-mediated effect comes from lymphocytic infiltrate on liver biopsy and lymphocytic colitis in idelalisib-treated patients. Additional evidence comes from the fact that the toxicity is both treatable and preventable with steroids, he said.
He cautioned that hepatotoxicity can recur rapidly when the drug is reintroduced.
“In general, our experience has been if idelalisib is resumed while the subject remains on steroids, the drug is more likely to be tolerated and the subject eventually can be tapered off steroids,” he said.
Asked by an audience member whether patients who are receiving idelalisib in the first-line setting should also receive steroids, Dr. Lampson said that they closely monitor patient liver enzymes around 28 days, and if grade 1 transaminitis is detected, patients are automatically started on low-dose steroids.
The study is sponsored by the Dana-Farber Cancer Institute in collaboration with Gilead Sciences and GlaxoSmithKline. Dr. Lampson and colleagues declared no relevant conflicts of interest.
AT ASH 2015
<p><b>Key clinical point: </b>Idelalisib in the first-line setting is associated with significant risk of hepatotoxicity, with a peak incidence at about 28 days of therapy.
</p><p><b>Major finding: </b>More than half (52%) of patients with newly diagnosed chronic lymphocytic leukemia had grade 3 or 4 hepatotoxicity with idelalisib monotherapy.
</p><p><b>Data source: </b>Ongoing phase II clinical trial with data on 24 patients.
</p><p><b>Disclosures: </b>The study is sponsored by the Dana-Farber Cancer Institute in collaboration with Gilead Sciences and GlaxoSmithKline. Dr. Lampson and colleagues declared no relevant conflicts of interest.</p>
